

Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades
- PMID: 14963156
- PMCID: PMC369207
- DOI: 10.1128/jvi.78.5.2537-2544.2004
Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades
Abstract
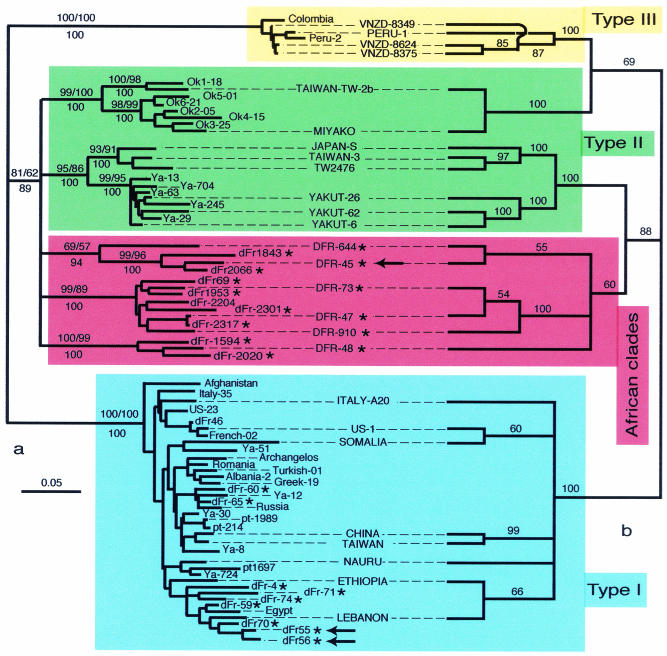
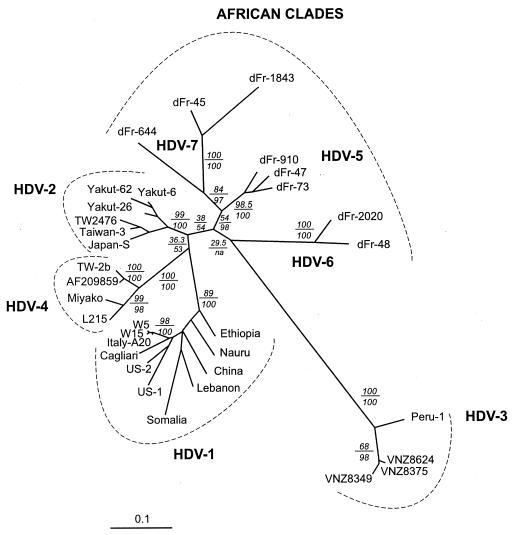
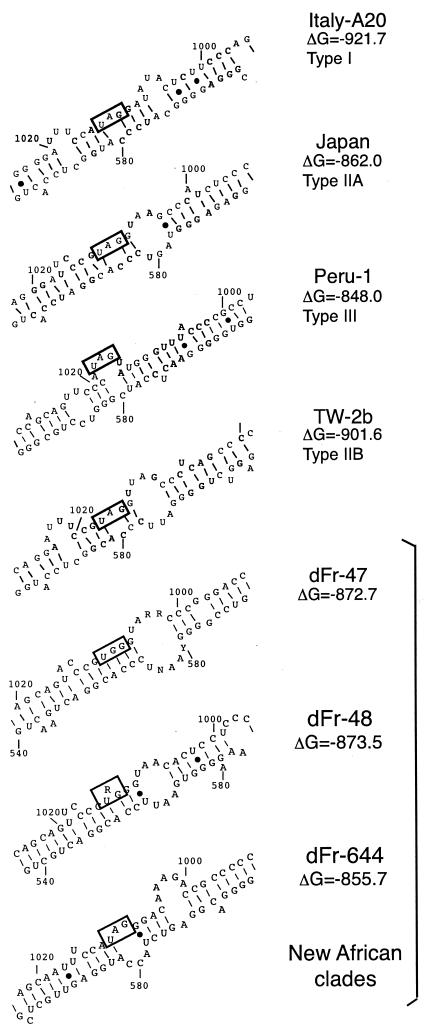
Hepatitis D virus (HDV) is a satellite of hepatitis B virus (HBV) for transmission and propagation and infects nearly 20 million people worldwide. The HDV genome is a compact circular single-stranded RNA genome with extensive intramolecular complementarity. Despite its different epidemiological and pathological patterns, the variability and geographical distribution of HDV are limited to three genotypes and two subtypes that have been characterized to date. Phylogenetic reconstructions based on the delta antigen gene and full-length genome sequence data show an extensive and probably ancient radiation of African lineages, suggesting that the genetic variability of HDV is much more complex than was previously thought, with evidence of additional clades. These results relate the geographic distribution of HDV more closely to the genetic variability of its helper HBV.
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
