

Incorporating genotyping uncertainty in haplotype inference for single-nucleotide polymorphisms
- PMID: 14966673
- PMCID: PMC1182263
- DOI: 10.1086/382284
Incorporating genotyping uncertainty in haplotype inference for single-nucleotide polymorphisms
Abstract
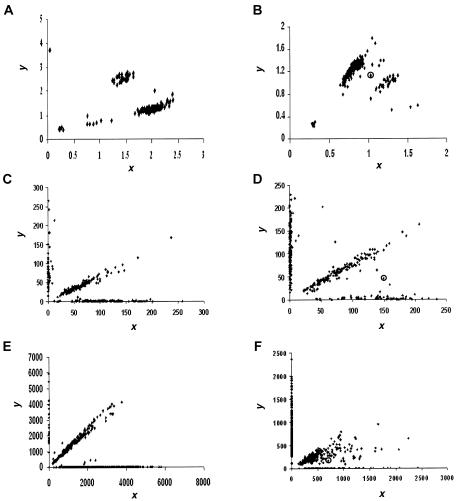
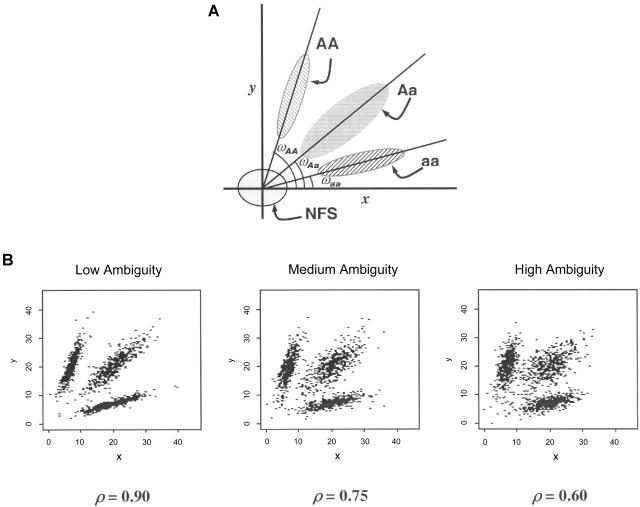
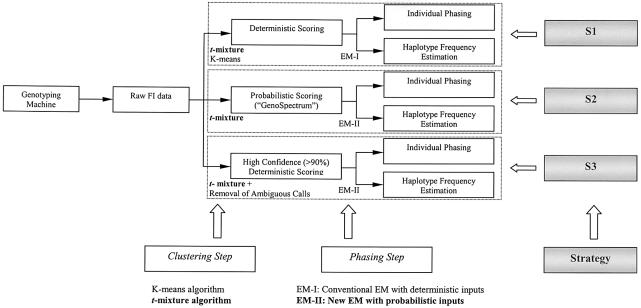
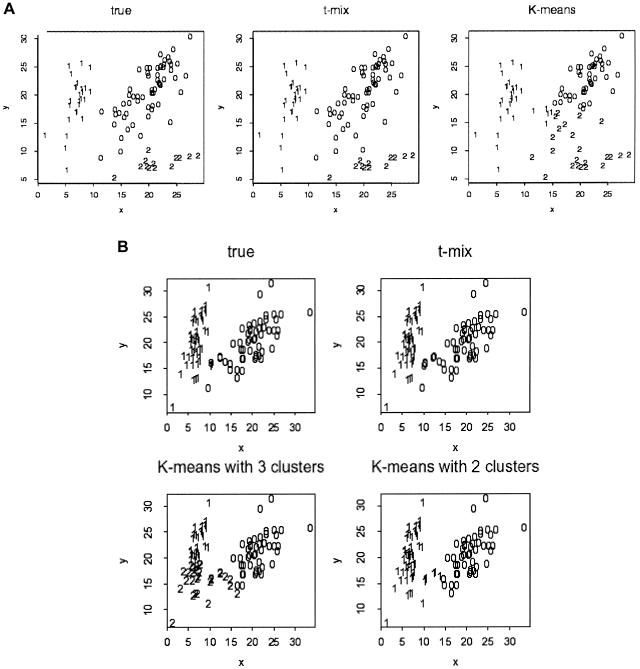
The accuracy of the vast amount of genotypic information generated by high-throughput genotyping technologies is crucial in haplotype analyses and linkage-disequilibrium mapping for complex diseases. To date, most automated programs lack quality measures for the allele calls; therefore, human interventions, which are both labor intensive and error prone, have to be performed. Here, we propose a novel genotype clustering algorithm, GeneScore, based on a bivariate t-mixture model, which assigns a set of probabilities for each data point belonging to the candidate genotype clusters. Furthermore, we describe an expectation-maximization (EM) algorithm for haplotype phasing, GenoSpectrum (GS)-EM, which can use probabilistic multilocus genotype matrices (called "GenoSpectrum") as inputs. Combining these two model-based algorithms, we can perform haplotype inference directly on raw readouts from a genotyping machine, such as the TaqMan assay. By using both simulated and real data sets, we demonstrate the advantages of our probabilistic approach over the current genotype scoring methods, in terms of both the accuracy of haplotype inference and the statistical power of haplotype-based association analyses.
Figures
References
Electronic-Database Information
-
- Authors' Web site, http://www.people.fas.harvard.edu/~junliu/genotype/ (for the GeneScore [probabilistic genotype clustering method using the t-mixture model] and GS-EM [the EM algorithm for haplotype phasing with multilocus GenoSpectrum inputs] software packages, their detailed instructions, and sample input and output files)
References
-
- Abecasis GR, Cherny SS, Cardon LR (2001) The impact of genotyping error on family-based analysis of quantitative traits. Eur J Hum Genet 9:130–134 - PubMed
-
- Akula N, Chen YS, Hennessy K, Schulze TG, Singh G, McMahon FJ (2002) Utility and accuracy of template-directed dye-terminator incorporation with fluorescence-polarization detection for genotyping single nucleotide polymorphisms. Biotechniques 32:1072–1078 - PubMed
-
- Clark AG (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 7:111–122 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
