

A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro
- PMID: 14973201
- PMCID: PMC373405
- DOI: 10.1093/nar/gkh271
A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro
Abstract
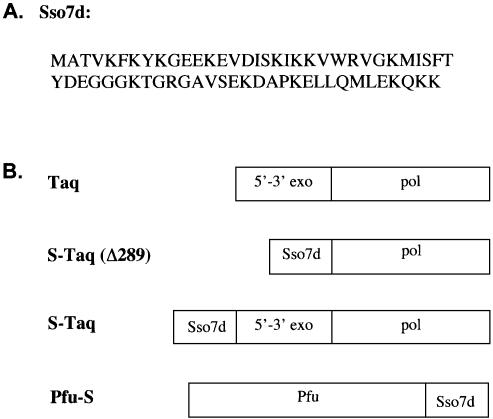
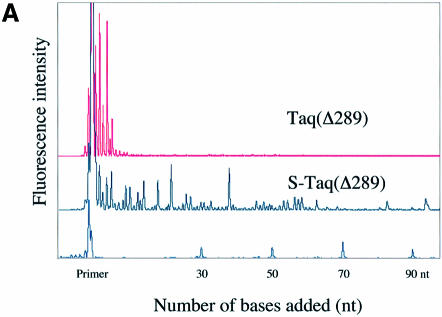
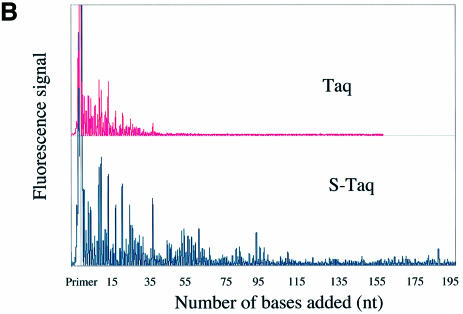
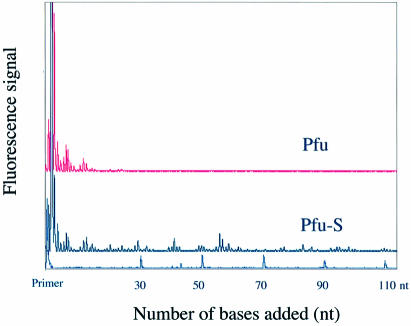
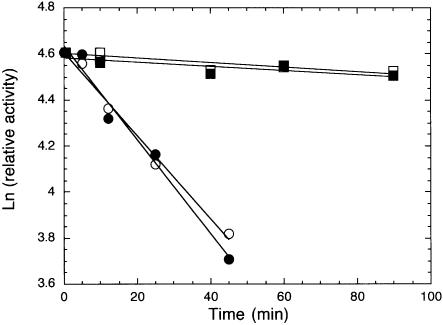
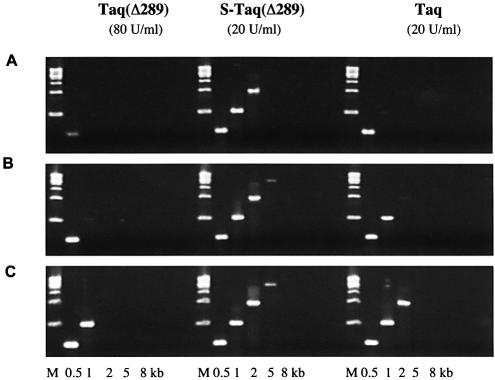
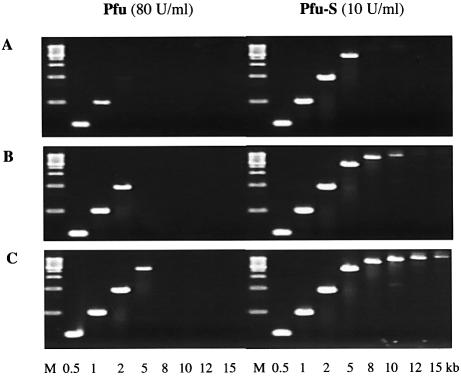
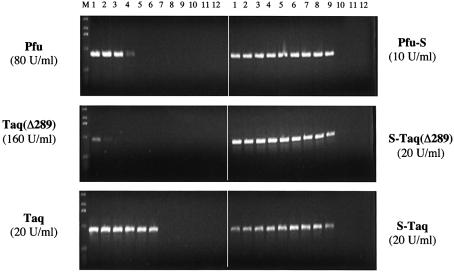
Mechanisms that allow replicative DNA polymerases to attain high processivity are often specific to a given polymerase and cannot be generalized to others. Here we report a protein engineering-based approach to significantly improve the processivity of DNA polymerases by covalently linking the polymerase domain to a sequence non-specific dsDNA binding protein. Using Sso7d from Sulfolobus solfataricus as the DNA binding protein, we demonstrate that the processivity of both family A and family B polymerases can be significantly enhanced. By introducing point mutations in Sso7d, we show that the dsDNA binding property of Sso7d is essential for the enhancement. We present evidence supporting two novel conclusions. First, the fusion of a heterologous dsDNA binding protein to a polymerase can increase processivity without compromising catalytic activity and enzyme stability. Second, polymerase processivity is limiting for the efficiency of PCR, such that the fusion enzymes exhibit profound advantages over unmodified enzymes in PCR applications. This technology has the potential to broadly improve the performance of nucleic acid modifying enzymes.
Figures
References
-
- Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd edn. W.H.Freeman, New York, NY.
-
- Nakai H., Beauchamp,B.B., Bernstein,J., Huber,H.E., Tabor,S. and Richardson,C.C. (1988) Formation and Propagation of the Bacteriophage T7 Replication Fork. American Society of Microbiology, Washington, DC.
-
- Bambara R.A., Uyemura,D. and Choi,T. (1978) On the processive mechanism of Escherichia coli DNA polymerase I. Quantitative assessment of processivity. J. Biol. Chem., 253, 413–423. - PubMed
-
- Das S.K. and Fujimura,R.K. (1977) Mechanism of T5-induced DNA polymerase. I. Replication of short primer templates. J. Biol. Chem., 252, 8700–8707. - PubMed
-
- Das S.K. and Fujimura,R.K. (1977) Mechanism of T5-induced DNA polymerase. II. Characterization of the dead-end complex. J. Biol. Chem., 252, 8708–8712. - PubMed
