

LSimpute: accurate estimation of missing values in microarray data with least squares methods
- PMID: 14978222
- PMCID: PMC374359
- DOI: 10.1093/nar/gnh026
LSimpute: accurate estimation of missing values in microarray data with least squares methods
Abstract
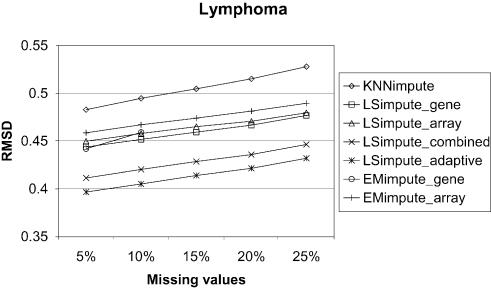
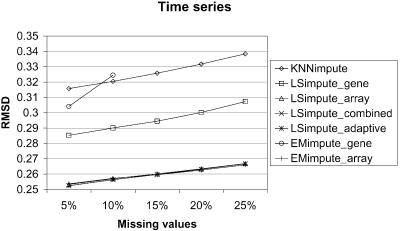
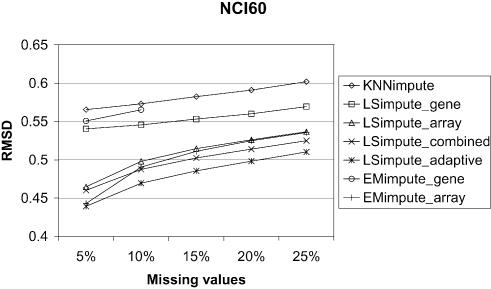
Microarray experiments generate data sets with information on the expression levels of thousands of genes in a set of biological samples. Unfortunately, such experiments often produce multiple missing expression values, normally due to various experimental problems. As many algorithms for gene expression analysis require a complete data matrix as input, the missing values have to be estimated in order to analyze the available data. Alternatively, genes and arrays can be removed until no missing values remain. However, for genes or arrays with only a small number of missing values, it is desirable to impute those values. For the subsequent analysis to be as informative as possible, it is essential that the estimates for the missing gene expression values are accurate. A small amount of badly estimated missing values in the data might be enough for clustering methods, such as hierachical clustering or K-means clustering, to produce misleading results. Thus, accurate methods for missing value estimation are needed. We present novel methods for estimation of missing values in microarray data sets that are based on the least squares principle, and that utilize correlations between both genes and arrays. For this set of methods, we use the common reference name LSimpute. We compare the estimation accuracy of our methods with the widely used KNNimpute on three complete data matrices from public data sets by randomly knocking out data (labeling as missing). From these tests, we conclude that our LSimpute methods produce estimates that consistently are more accurate than those obtained using KNNimpute. Additionally, we examine a more classic approach to missing value estimation based on expectation maximization (EM). We refer to our EM implementations as EMimpute, and the estimate errors using the EMimpute methods are compared with those our novel methods produce. The results indicate that on average, the estimates from our best performing LSimpute method are at least as accurate as those from the best EMimpute algorithm.
Figures
References
-
- Perou C.M., Sørlie,T., Eisen,M.B., van de Rijn,M., Jeffrey,S.S., Rees,C.A., Pollack,J.R., Ross,D.T., Johnsen,H., Akslen,L.A. et al. (2000) Molecular portraits of human breast tumors. Nature, 406, 747–752. - PubMed
-
- Alizadeh A.A., Eisen,M.B., Davis,R.E., Ma,C., Lossos,I.S., Rosenwald,A., Boldrick,J.C., Sabet,H., Tran, T, Powell,J.L. et al. (2000) Distinct types of diffuse large B-cell lymphoma identified by gene-expression profiling. Nature, 403, 503–511. - PubMed
-
- Golub T.R., Slonim,D.K., Tamayo,P., Huard,C., Gaasenbeeck,M., Mesirov,J.P., Coller,H., Loh,M.L., Downing,J.R., Caligiuri,M.A. et al. (1999) Molecular classification of cancer: class discovery and class prediction by expression monitoring. Science, 286, 531–537. - PubMed
-
- Chu S., DeRisi,J., Eisen,M.B., Mulholland,J., Botstein,D., Brown,P.O. and Hesrkowitz,I. (1998) The transcriptional program of sporulation in budding yeast. Science, 278, 680–686. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
