

Improved side-chain prediction accuracy using an ab initio potential energy function and a very large rotamer library
- PMID: 14978310
- PMCID: PMC2286725
- DOI: 10.1110/ps.03250104
Improved side-chain prediction accuracy using an ab initio potential energy function and a very large rotamer library
Abstract
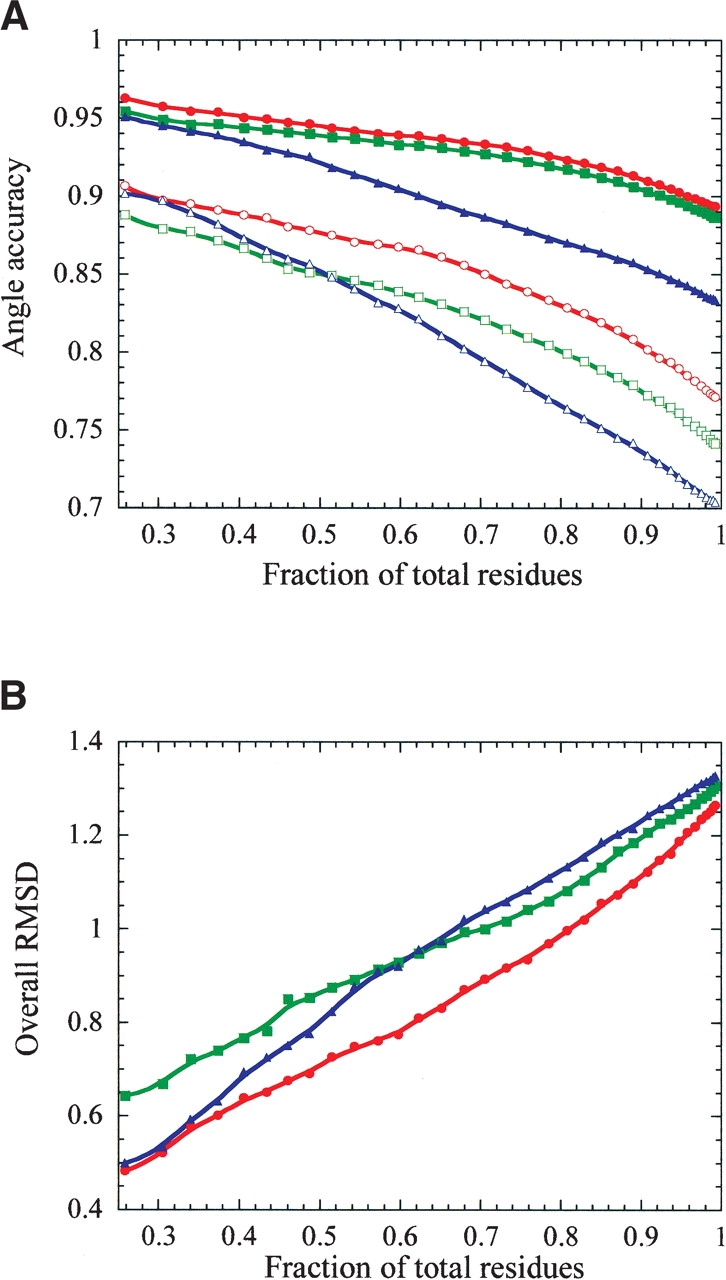
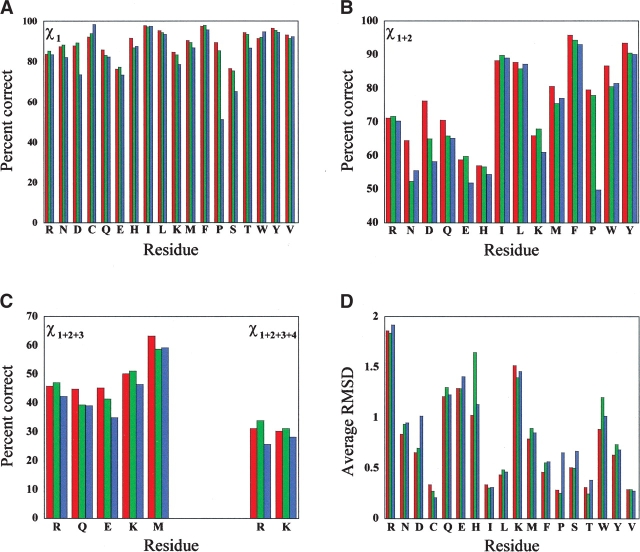
Accurate prediction of the placement and comformations of protein side chains given only the backbone trace has a wide range of uses in protein design, structure prediction, and functional analysis. Prediction has most often relied on discrete rotamer libraries so that rapid fitness of side-chain rotamers can be assessed against some scoring function. Scoring functions are generally based on experimental parameters from small-molecule studies or empirical parameters based on determined protein structures. Here, we describe the NCN algorithm for predicting the placement of side chains. A predominantly first-principles approach was taken to develop the potential energy function incorporating van der Waals and electrostatics based on the OPLS parameters, and a hydrogen bonding term. The only empirical knowledge used is the frequency of rotameric states from the PDB. The rotamer library includes nearly 50,000 rotamers, and is the most extensive discrete library used to date. Although the computational time tends to be longer than most other algorithms, the overall accuracy exceeds all algorithms in the literature when placing rotamers on an accurate backbone trace. Considering only the most buried residues, 80% of the total residues tested, the placement accuracy reaches 92% for chi(1), and 83% for chi(1 + 2), and an overall RMS deviation of 1 A. Additionally, we show that if information is available to restrict chi(1) to one rotamer well, then this algorithm can generate structures with an average RMS deviation of 1.0 A for all heavy side-chains atoms and a corresponding overall chi(1 + 2) accuracy of 85.0%.
Figures
Similar articles
-
A protein-dependent side-chain rotamer library.BMC Bioinformatics. 2011 Dec 14;12 Suppl 14(Suppl 14):S10. doi: 10.1186/1471-2105-12-S14-S10. BMC Bioinformatics. 2011. PMID: 22373394 Free PMC article.
-
Side-chain modeling with an optimized scoring function.Protein Sci. 2002 Feb;11(2):322-31. doi: 10.1110/ps.24902. Protein Sci. 2002. PMID: 11790842 Free PMC article.
-
Incorporating knowledge-based biases into an energy-based side-chain modeling method: application to comparative modeling of protein structure.Biopolymers. 2001 Aug;59(2):72-86. doi: 10.1002/1097-0282(200108)59:2<72::AID-BIP1007>3.0.CO;2-S. Biopolymers. 2001. PMID: 11373721
-
Rotamer Dynamics: Analysis of Rotamers in Molecular Dynamics Simulations of Proteins.Biophys J. 2019 Jun 4;116(11):2062-2072. doi: 10.1016/j.bpj.2019.04.017. Epub 2019 Apr 22. Biophys J. 2019. PMID: 31084902 Free PMC article. Review.
-
Rotamer libraries in the 21st century.Curr Opin Struct Biol. 2002 Aug;12(4):431-40. doi: 10.1016/s0959-440x(02)00344-5. Curr Opin Struct Biol. 2002. PMID: 12163064 Review.
Cited by
-
MCCE2: improving protein pKa calculations with extensive side chain rotamer sampling.J Comput Chem. 2009 Nov 15;30(14):2231-47. doi: 10.1002/jcc.21222. J Comput Chem. 2009. PMID: 19274707 Free PMC article.
-
A protein-dependent side-chain rotamer library.BMC Bioinformatics. 2011 Dec 14;12 Suppl 14(Suppl 14):S10. doi: 10.1186/1471-2105-12-S14-S10. BMC Bioinformatics. 2011. PMID: 22373394 Free PMC article.
-
FASPR: an open-source tool for fast and accurate protein side-chain packing.Bioinformatics. 2020 Jun 1;36(12):3758-3765. doi: 10.1093/bioinformatics/btaa234. Bioinformatics. 2020. PMID: 32259206 Free PMC article.
-
Explicit orientation dependence in empirical potentials and its significance to side-chain modeling.Acc Chem Res. 2009 Aug 18;42(8):1087-96. doi: 10.1021/ar900009e. Acc Chem Res. 2009. PMID: 19445451 Free PMC article.
-
Modeling mutations in protein structures.Protein Sci. 2007 Sep;16(9):2030-41. doi: 10.1110/ps.072855507. Protein Sci. 2007. PMID: 17766392 Free PMC article.
References
-
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217.
-
- Chothia, C. 1976. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 105 1–14. - PubMed
-
- Claussen, H., Buning, C., Rarey, M., and Lengauer, T. 2001. FlexE: Efficient molecular docking considering protein structure variations. J. Mol. Biol. 308 377–395. - PubMed
-
- Dahiyat, B.I. and Mayo, S.L. 1997. De novo protein design: Fully automated sequence selection. Science 278 82–87. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
