

Sensitivity and selectivity in protein structure comparison
- PMID: 14978311
- PMCID: PMC2286722
- DOI: 10.1110/ps.03328504
Sensitivity and selectivity in protein structure comparison
Abstract
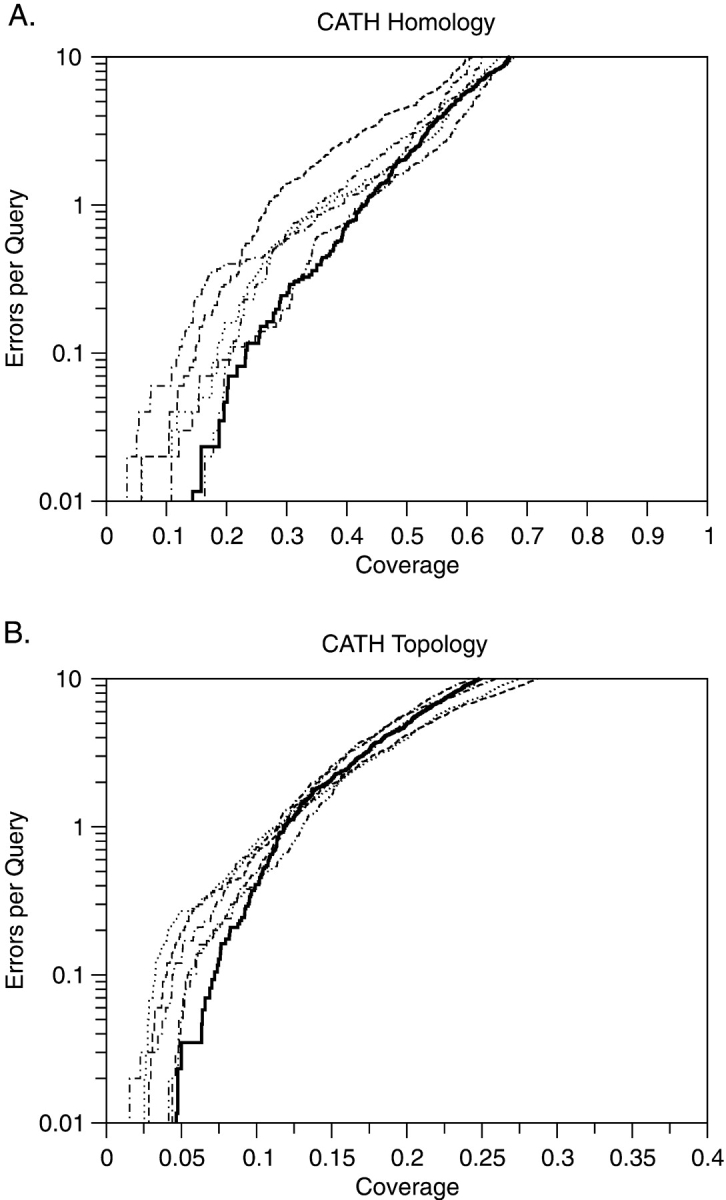
Seven protein structure comparison methods and two sequence comparison programs were evaluated on their ability to detect either protein homologs or domains with the same topology (fold) as defined by the CATH structure database. The structure alignment programs Dali, Structal, Combinatorial Extension (CE), VAST, and Matras were tested along with SGM and PRIDE, which calculate a structural distance between two domains without aligning them. We also tested two sequence alignment programs, SSEARCH and PSI-BLAST. Depending upon the level of selectivity and error model, structure alignment programs can detect roughly twice as many homologous domains in CATH as sequence alignment programs. Dali finds the most homologs, 321-533 of 1120 possible true positives (28.7%-45.7%), at an error rate of 0.1 errors per query (EPQ), whereas PSI-BLAST finds 365 true positives (32.6%), regardless of the error model. At an EPQ of 1.0, Dali finds 42%-70% of possible homologs, whereas Matras finds 49%-57%; PSI-BLAST finds 36.9%. However, Dali achieves >84% coverage before the first error for half of the families tested. Dali and PSI-BLAST find 9.2% and 5.2%, respectively, of the 7056 possible topology pairs at an EPQ of 0.1 and 19.5, and 5.9% at an EPQ of 1.0. Most statistical significance estimates reported by the structural alignment programs overestimate the significance of an alignment by orders of magnitude when compared with the actual distribution of errors. These results help quantify the statistical distinction between analogous and homologous structures, and provide a benchmark for structure comparison statistics.
Figures




References
-
- Altschul, S.F. and Gish, W. 1996. Local alignment statistics. Methods Enzymol. 266 460–480. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
