

Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice
- PMID: 14985425
- PMCID: PMC6730389
- DOI: 10.1523/JNEUROSCI.4406-03.2004
Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice
Abstract
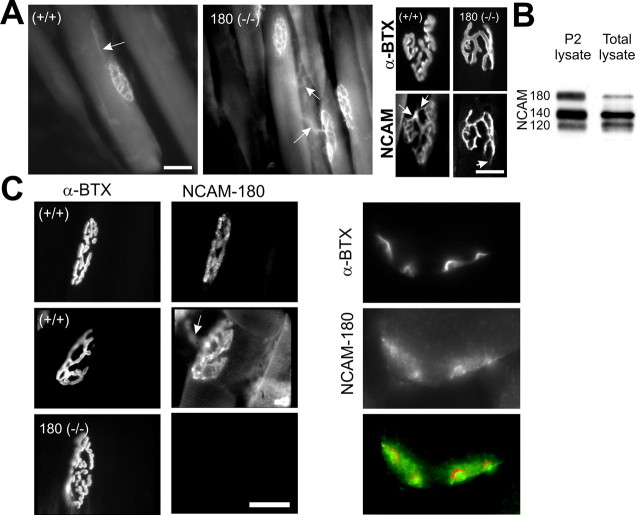
Mice that lack all three major isoforms of neural cell adhesion molecule (NCAM) (180 and 140 kDa transmembrane, and 120 kDa glycosylphosphatidylinositol linked) were previously shown to exhibit major alterations in the maturation of their neuromuscular junctions (NMJs). Specifically, even by postnatal day 30, they failed to downregulate from along their axons and terminals an immature, brefeldin A-sensitive, synaptic vesicle-cycling mechanism that used L-type Ca2+ channels. In addition, these NCAM null NMJs were unable to maintain effective transmitter output with high-frequency repetitive stimulation, exhibiting both severe initial depression and subsequent cyclical periods of total transmission failures that were of presynaptic origin. As reported here, mice that lack only the 180 kDa isoform of NCAM downregulated the immature vesicle-cycling mechanism on schedule, implicating either the 140 or 120 kDa NCAM isoforms in this important maturational event. However, 180 NCAM-deficient mice still exhibited many functional transmission defects. Although 180 NCAM null NMJs did not show the severe initial depression of NCAM null NMJs, they still had cyclical periods of complete transmission failure. In addition, several presynaptic molecules were expressed at lower levels or were more diffusely localized. Thus, the 180 kDa isoform of NCAM appears to play an important role in the molecular organization of the presynaptic terminal and in ensuring effective transmitter output with repetitive stimulation. Our results also suggest that PKC and MLCK (myosin light chain kinase) may be downstream effectors of NCAM in these processes. Together, these results indicate that different isoforms of NCAM mediate distinct and important events in presynaptic maturation.
Figures








References
-
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N (1998) Munc 13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron 21: 123-136. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous