

Paradigms for computational nucleic acid design
- PMID: 14990744
- PMCID: PMC390280
- DOI: 10.1093/nar/gkh291
Paradigms for computational nucleic acid design
Abstract
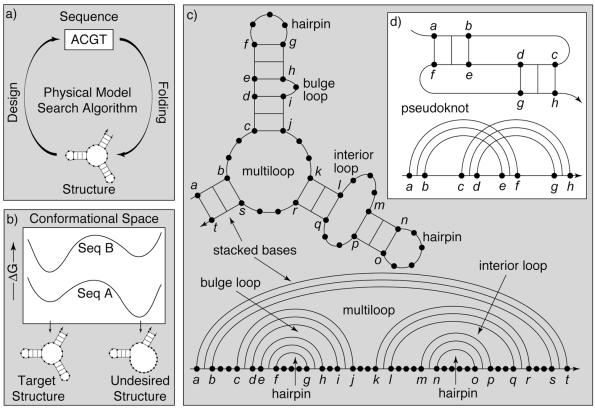
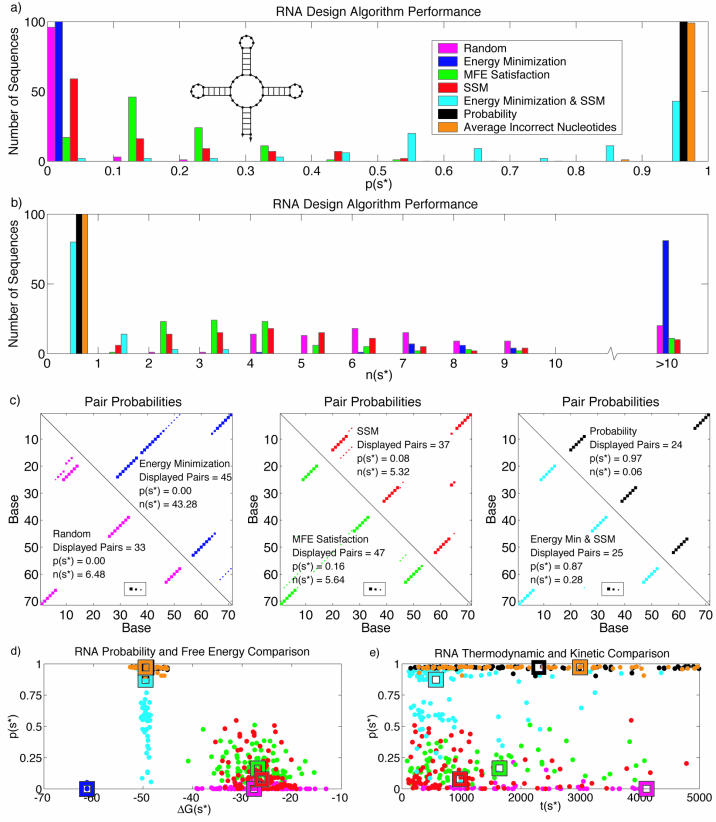
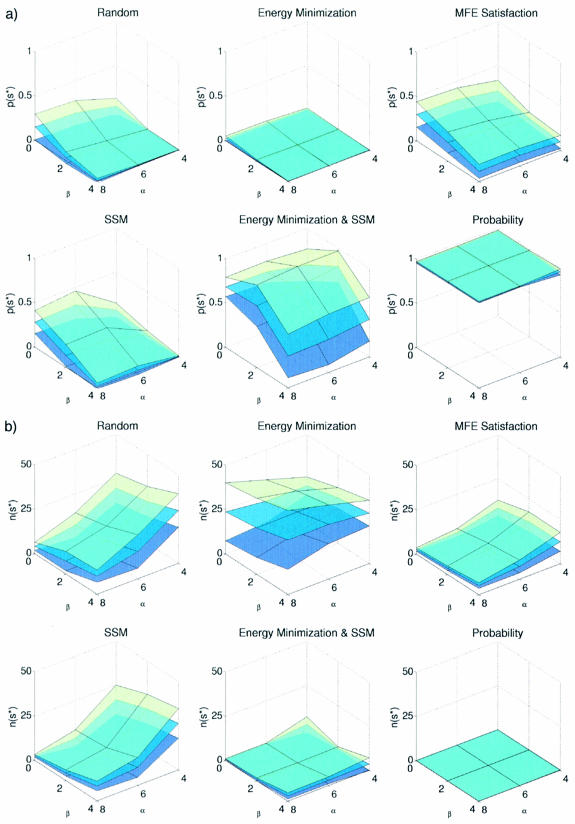
The design of DNA and RNA sequences is critical for many endeavors, from DNA nanotechnology, to PCR-based applications, to DNA hybridization arrays. Results in the literature rely on a wide variety of design criteria adapted to the particular requirements of each application. Using an extensively studied thermodynamic model, we perform a detailed study of several criteria for designing sequences intended to adopt a target secondary structure. We conclude that superior design methods should explicitly implement both a positive design paradigm (optimize affinity for the target structure) and a negative design paradigm (optimize specificity for the target structure). The commonly used approaches of sequence symmetry minimization and minimum free-energy satisfaction primarily implement negative design and can be strengthened by introducing a positive design component. Surprisingly, our findings hold for a wide range of secondary structures and are robust to modest perturbation of the thermodynamic parameters used for evaluating sequence quality, suggesting the feasibility and ongoing utility of a unified approach to nucleic acid design as parameter sets are refined further. Finally, we observe that designing for thermodynamic stability does not determine folding kinetics, emphasizing the opportunity for extending design criteria to target kinetic features of the energy landscape.
Figures



References
-
- Seeman N.C. (1982) Nucleic acid junctions and lattices. J. Theor. Biol., 99, 237–247. - PubMed
-
- Seeman N.C. (1999) DNA engineering and its application to nanotechnology. Trends Biotechnol., 17, 437–443. - PubMed
-
- Winfree E., Liu,F., Wenzler,L.A. and Seeman,N.C. (1998) Design and self-assembly of two-dimensional DNA crystals. Nature, 394, 539–544. - PubMed
-
- Kallenbach R.K., Ma,R.-I. and Seeman,N.C. (1983) An immobile nucleic acid junction constructed from oligonucleotides. Nature, 305, 829–831.
-
- Chen J. and Seeman,N.C. (1991) The synthesis from DNAs of a molecule with the connectivity of a cube. Nature, 350, 631–633. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
