

Codon usage between genomes is constrained by genome-wide mutational processes
- PMID: 14990797
- PMCID: PMC373487
- DOI: 10.1073/pnas.0307827100
Codon usage between genomes is constrained by genome-wide mutational processes
Abstract
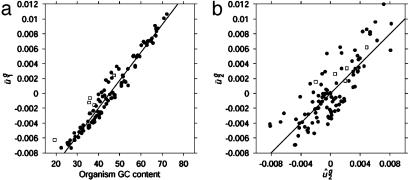
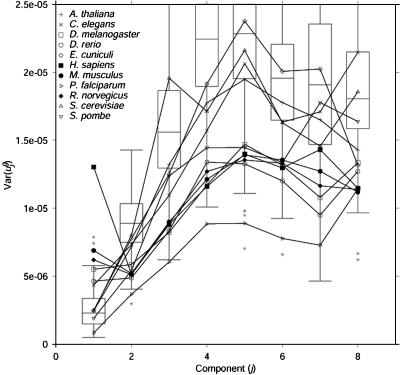
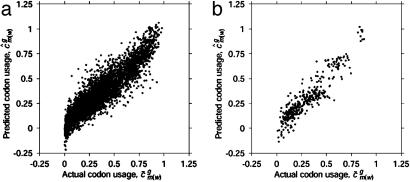
Analysis of genome-wide codon bias shows that only two parameters effectively differentiate the genome-wide codon bias of 100 eubacterial and archaeal organisms. The first parameter correlates with genome GC content, and the second parameter correlates with context-dependent nucleotide bias. Both of these parameters may be calculated from intergenic sequences. Therefore, genome-wide codon bias in eubacteria and archaea may be predicted from intergenic sequences that are not translated. When these two parameters are calculated for genes from nonmammalian eukaryotic organisms, genes from the same organism again have similar values, and genome-wide codon bias may also be predicted from intergenic sequences. In mammals, genes from the same organism are similar only in the second parameter, because GC content varies widely among isochores. Our results suggest that, in general, genome-wide codon bias is determined primarily by mutational processes that act throughout the genome, and only secondarily by selective forces acting on translated sequences.
Figures

References
-
- Grantham, R. (1980) Trends Biochem. Sci. 5, 327-331.
-
- Ikemura, T. (1985) Mol. Biol. Evol. 2, 13-34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
