

Increased CD36 protein as a response to defective insulin signaling in macrophages
- PMID: 14991075
- PMCID: PMC351316
- DOI: 10.1172/JCI19528
Increased CD36 protein as a response to defective insulin signaling in macrophages
Abstract
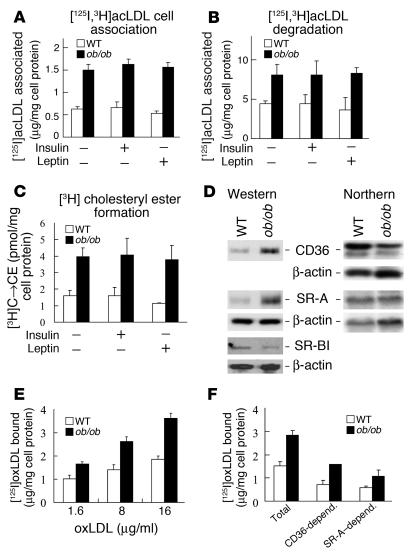
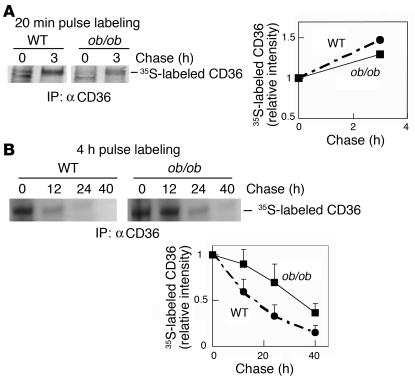
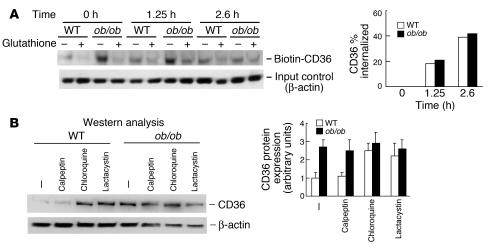
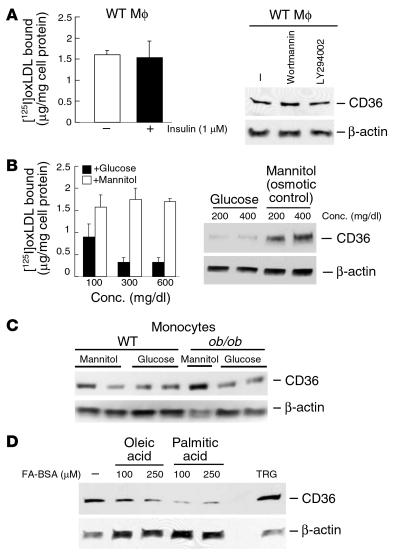
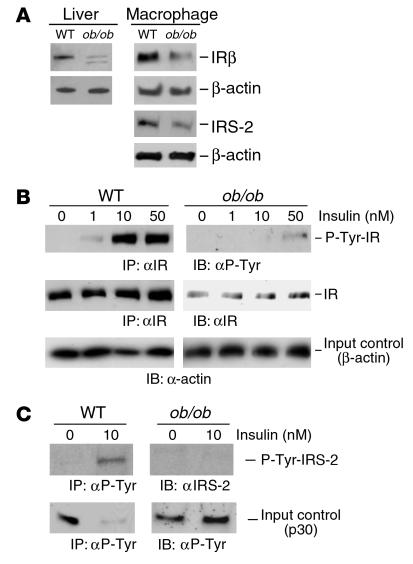
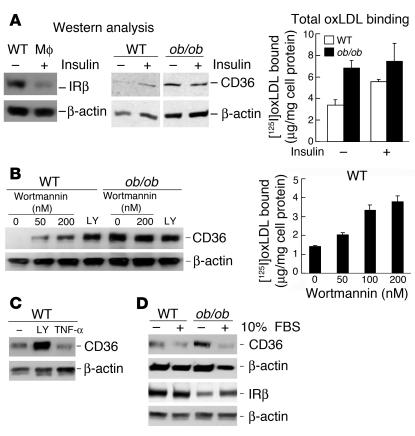
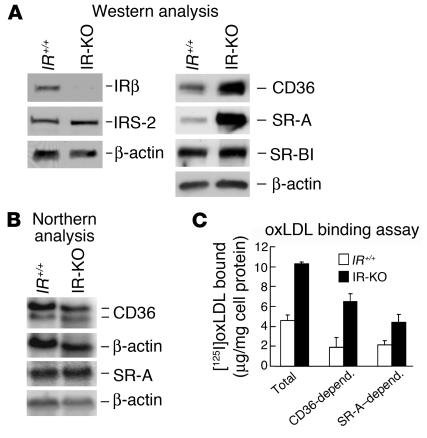
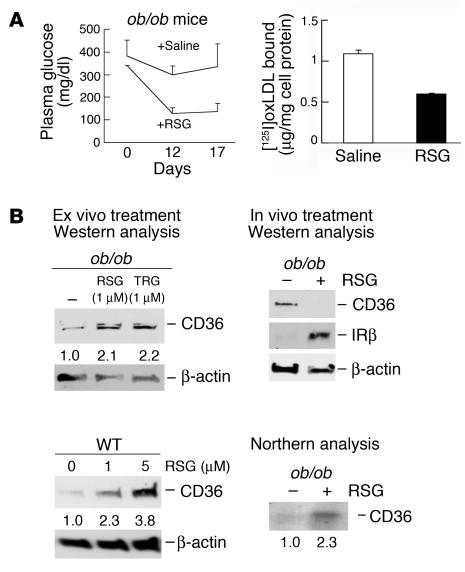
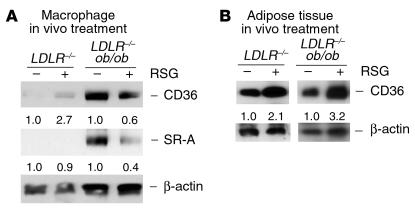
Accelerated atherosclerosis is a major cause of morbidity and death in insulin-resistant states such as obesity and the metabolic syndrome, but the underlying mechanisms are poorly understood. We show that macrophages from obese (ob/ob) mice have increased binding and uptake of oxidized LDL, in part due to a post-transcriptional increase in CD36 protein. Macrophages from ob/ob mice are also insulin resistant, as shown by reduced expression and signaling of insulin receptors. Three lines of evidence indicate that the increase in CD36 is caused by defective insulin signaling: (a) Treatment of wild-type macrophages with LY294002, an inhibitor of insulin signaling via PI3K, results in an increase in CD36; (b) insulin receptor knockout macrophages show a post-transcriptional increase in CD36 protein; and (c) administration of thiazolidinediones to intact ob/ob mice and ob/ob, LDL receptor-deficient mice results in a reversal of macrophage insulin receptor defects and decreases CD36 protein. The last finding contrasts with the increase in CD36 that results from treatment of macrophages with these drugs ex vivo. The results suggest that defective macrophage insulin signaling predisposes to foam cell formation and atherosclerosis in insulin-resistant states and that this is reversed in vivo by treatment with PPAR-gamma activators.
Figures
References
-
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998;339:229–234. - PubMed
-
- Reaven GM. Multiple CHD risk factors in type 2 diabetes: beyond hyperglycaemia. Diabetes Obes. Metab. 2002;4:S13–S18. - PubMed
-
- Haffner SM. Lipoprotein disorders associated with type 2 diabetes mellitus and insulin resistance. Am. J. Cardiol. 2002;90:55i–61i. - PubMed
-
- Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45:623–634. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
