

Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22
- PMID: 14993201
- PMCID: PMC353210
- DOI: 10.1101/gr.2094104
Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22
Abstract
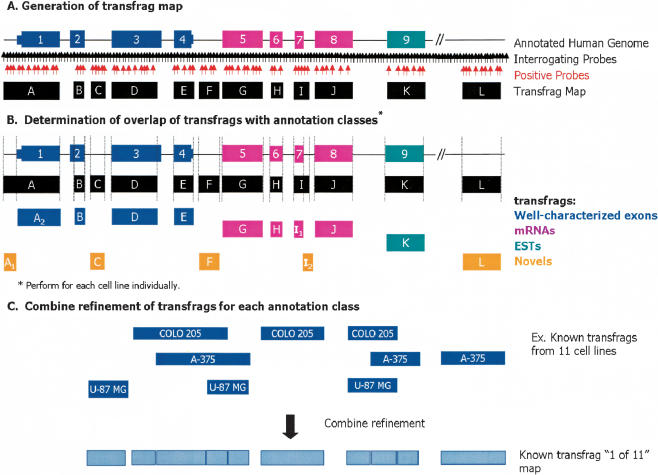
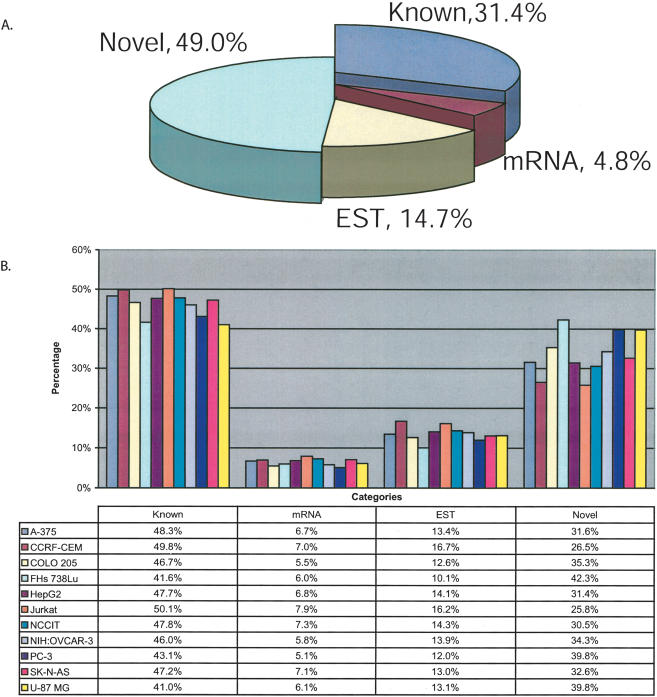
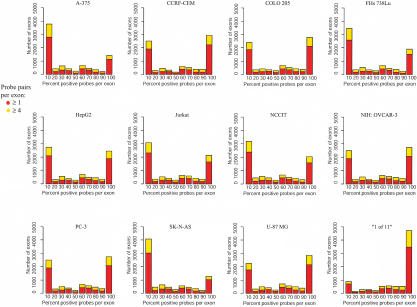
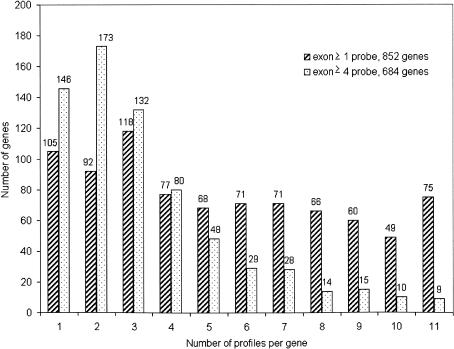
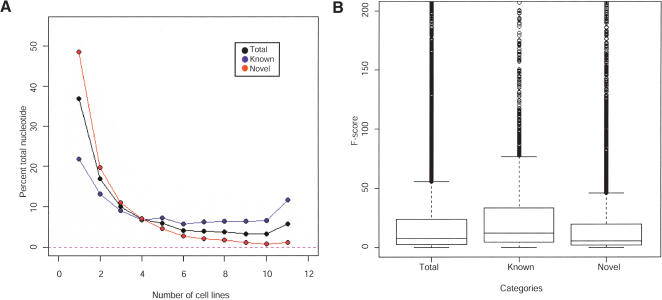
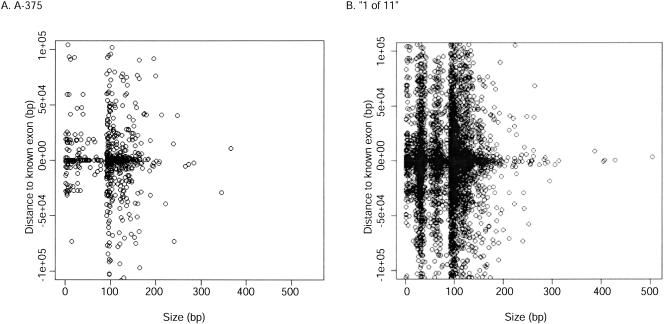
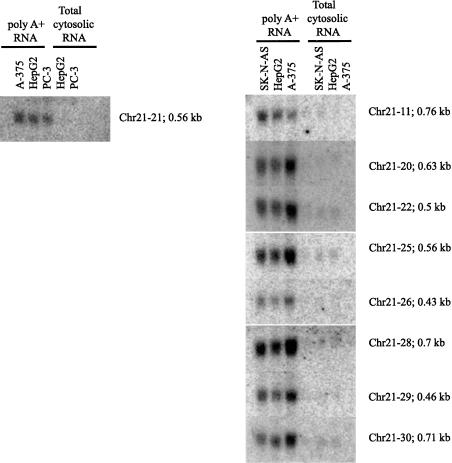
In this report, we have achieved a richer view of the transcriptome for Chromosomes 21 and 22 by using high-density oligonucleotide arrays on cytosolic poly(A)(+) RNA. Conservatively, only 31.4% of the observed transcribed nucleotides correspond to well-annotated genes, whereas an additional 4.8% and 14.7% correspond to mRNAs and ESTs, respectively. Approximately 85% of the known exons were detected, and up to 21% of known genes have only a single isoform based on exon-skipping alternative expression. Overall, the expression of the well-characterized exons falls predominately into two categories, uniquely or ubiquitously expressed with an identifiable proportion of antisense transcripts. The remaining observed transcription (49.0%) was outside of any known annotation. These novel transcripts appear to be more cell-line-specific and have lower and less variation in expression than the well-characterized genes. Novel transcripts were further characterized based on their distance to annotations, transcript size, coding capacity, and identification as antisense to intronic sequences. By RT-PCR, 126 novel transcripts were independently verified, resulting in a 65% verification rate. These observations strongly support the argument for a re-evaluation of the total number of human genes and an alternative term for "gene" to encompass these growing, novel classes of RNA transcripts in the human genome.
Figures
References
-
- Bolstad, B.M., Irizarry, R.A., Astrand, M., and Speed, T.P. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193. - PubMed
-
- Cawley, S., Bekiranov, S., Ng, H., Kapranov, P., Sekinger, E.A., Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A., et al. 2004. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 point to widespread regulation of non-coding RNAs. Cell (in press). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources