

Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands
- PMID: 14993236
- PMCID: PMC2172154
- DOI: 10.1083/jcb.200307137
Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands
Abstract
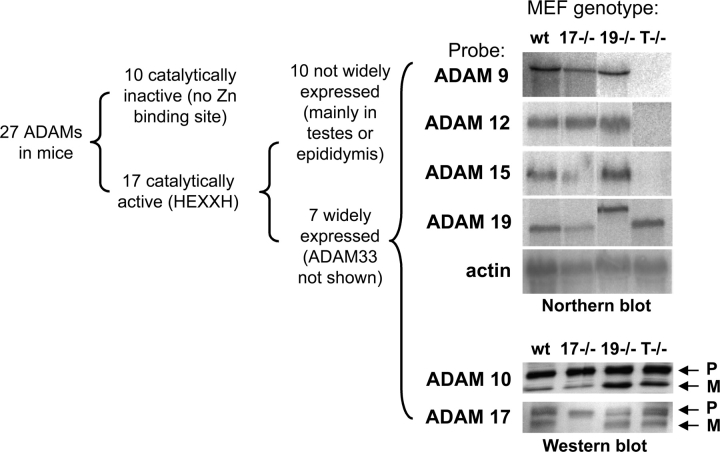
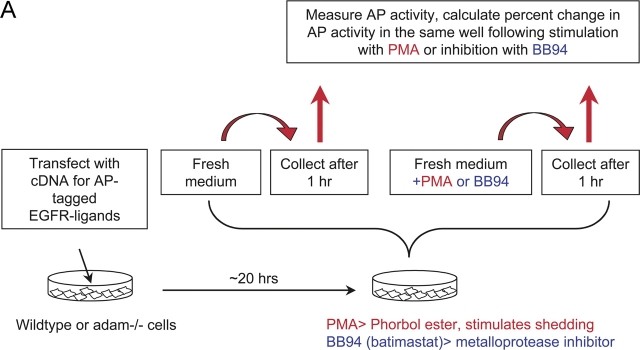
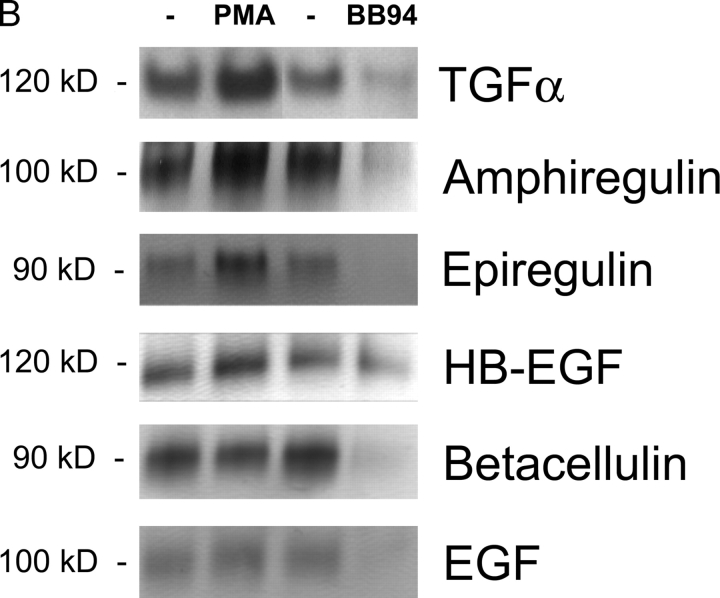
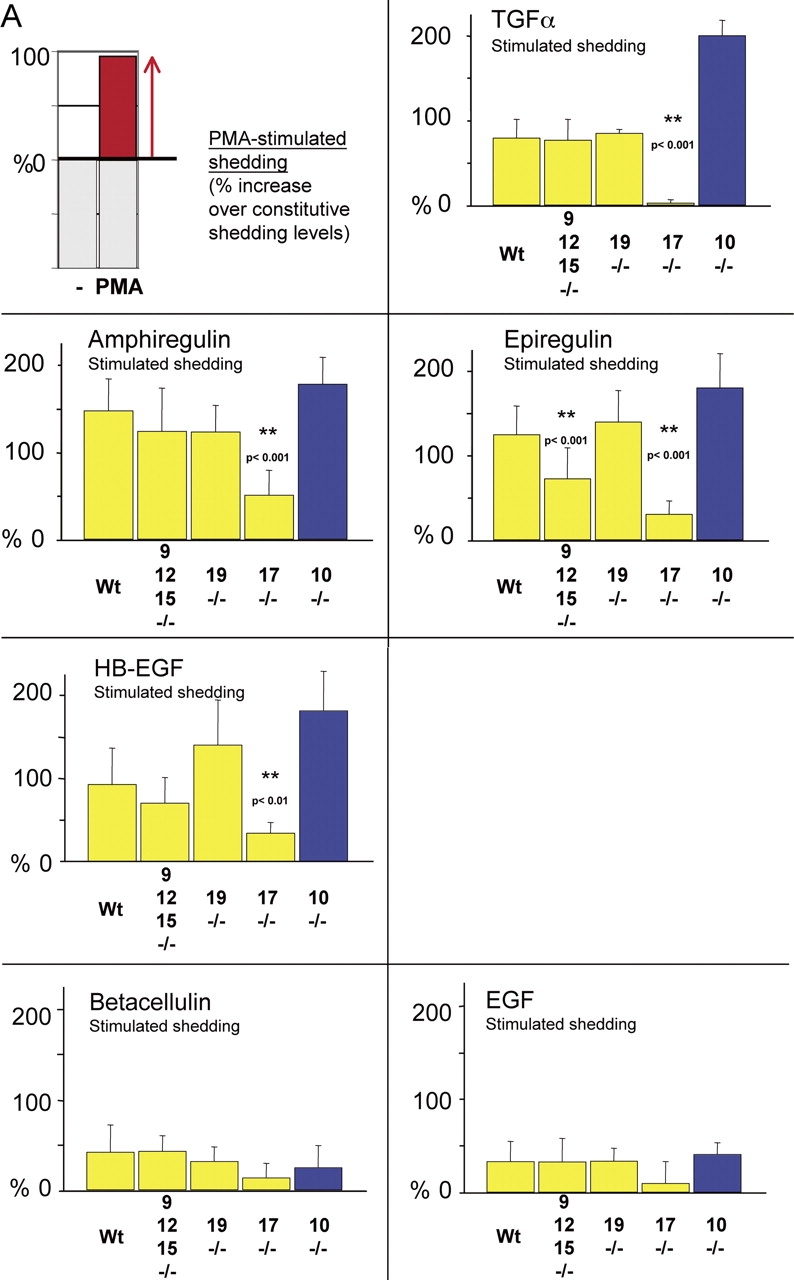
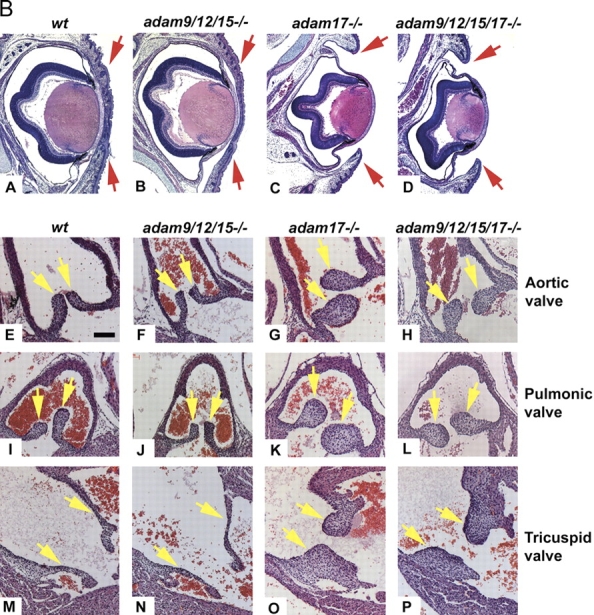
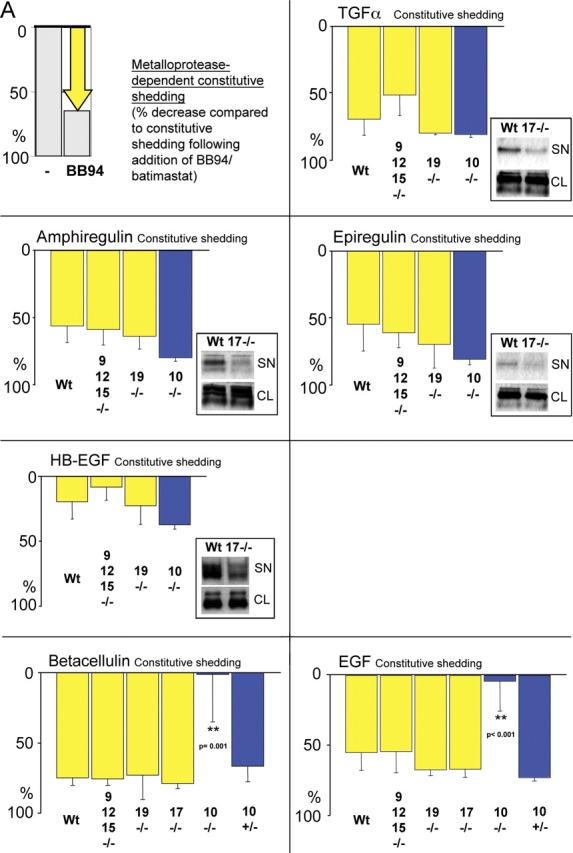
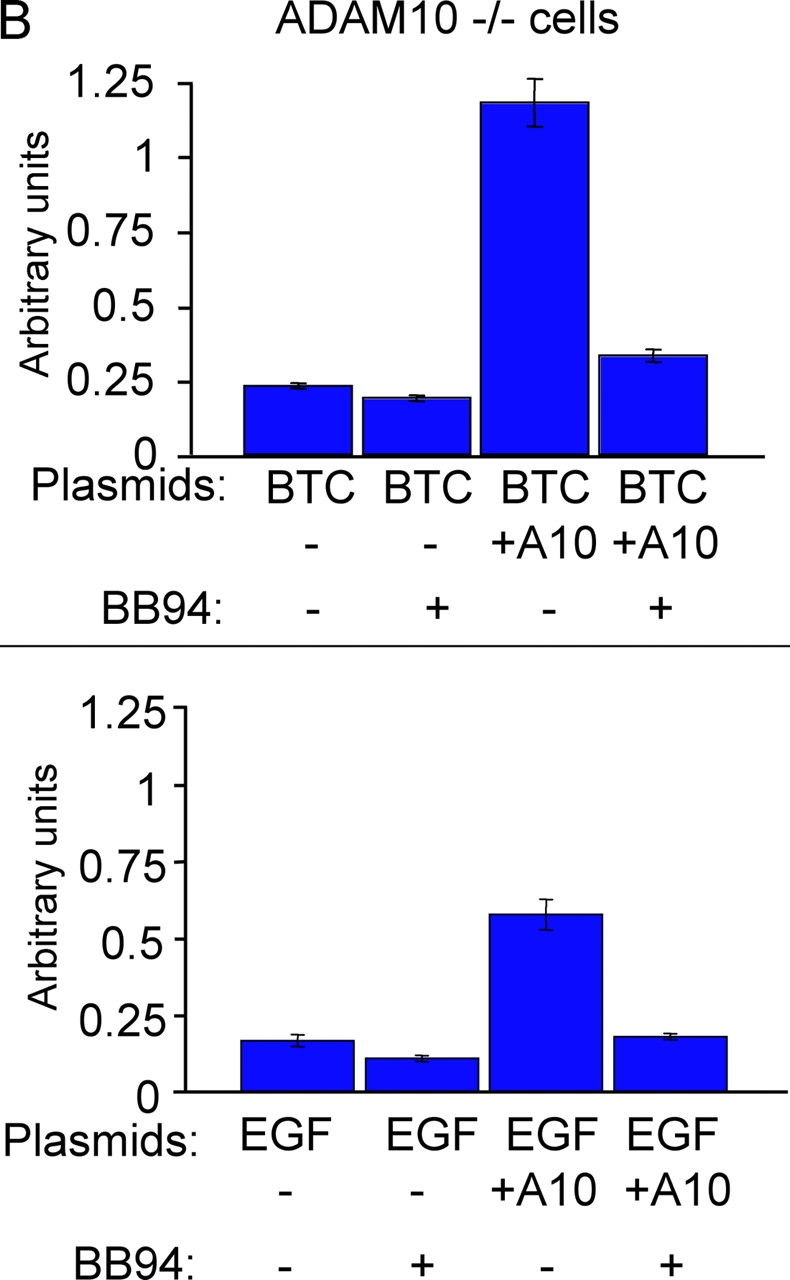
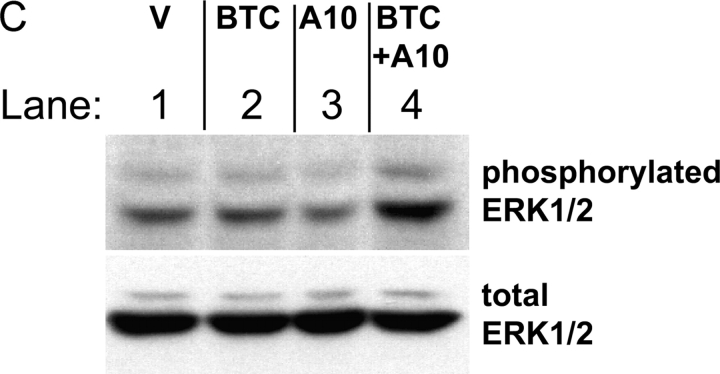
All ligands of the epidermal growth factor receptor (EGFR), which has important roles in development and disease, are released from the membrane by proteases. In several instances, ectodomain release is critical for activation of EGFR ligands, highlighting the importance of identifying EGFR ligand sheddases. Here, we uncovered the sheddases for six EGFR ligands using mouse embryonic cells lacking candidate-releasing enzymes (a disintegrin and metalloprotease [ADAM] 9, 10, 12, 15, 17, and 19). ADAM10 emerged as the main sheddase of EGF and betacellulin, and ADAM17 as the major convertase of epiregulin, transforming growth factor alpha, amphiregulin, and heparin-binding EGF-like growth factor in these cells. Analysis of adam9/12/15/17-/- knockout mice corroborated the essential role of adam17-/- in activating the EGFR in vivo. This comprehensive evaluation of EGFR ligand shedding in a defined experimental system demonstrates that ADAMs have critical roles in releasing all EGFR ligands tested here. Identification of EGFR ligand sheddases is a crucial step toward understanding the mechanism underlying ectodomain release, and has implications for designing novel inhibitors of EGFR-dependent tumors.
Figures
References
-
- Asakura, M., M. Kitakaze, S. Takashima, Y. Liao, F. Ishikura, T. Yoshinaka, H. Ohmoto, K. Node, K. Yoshino, H. Ishiguro, et al. 2002. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat. Med. 8:35–40. - PubMed
-
- Black, R.A., and J.M. White. 1998. ADAMs: focus on the protease domain. Curr. Opin. Cell Biol. 10:654–659. - PubMed
-
- Brachmann, R., P.B. Lindquist, M. Nagashima, W. Kohr, T. Lipari, M. Napier, and R. Derynck. 1989. Transmembrane TGF-α precursors activate EGF/TGF-α receptors. Cell. 56:691–700. - PubMed
-
- Chesneau, V., D. Becherer, Y. Zheng, H. Erdjument-Bromage, P. Tempst, and C.P. Blobel. 2003. Catalytic properties of ADAM19. J. Biol. Chem. 278:22331–22340. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
