

Inhibition of the Jun N-terminal protein kinase pathway by SHIP-1, a lipid phosphatase that interacts with the adaptor molecule Dok-3
- PMID: 14993273
- PMCID: PMC355862
- DOI: 10.1128/MCB.24.6.2332-2343.2004
Inhibition of the Jun N-terminal protein kinase pathway by SHIP-1, a lipid phosphatase that interacts with the adaptor molecule Dok-3
Abstract
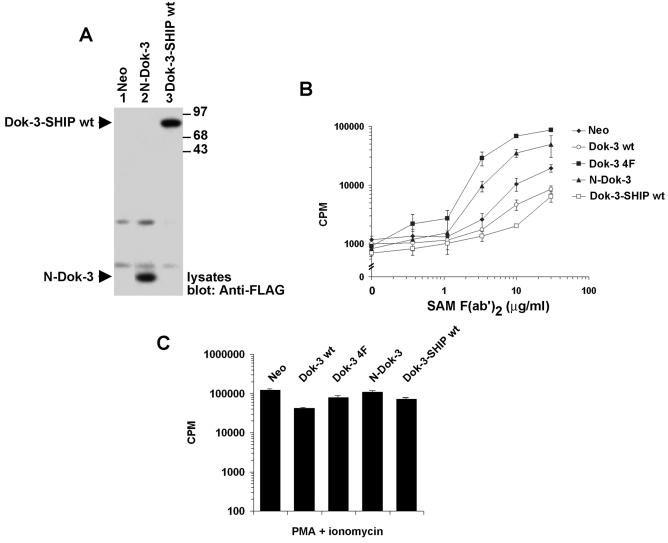
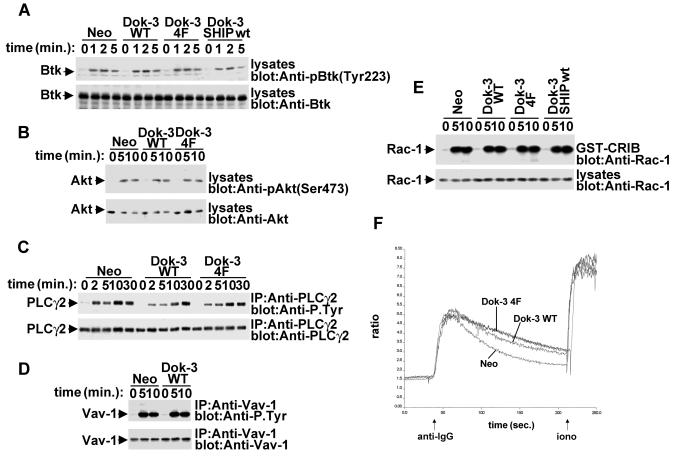
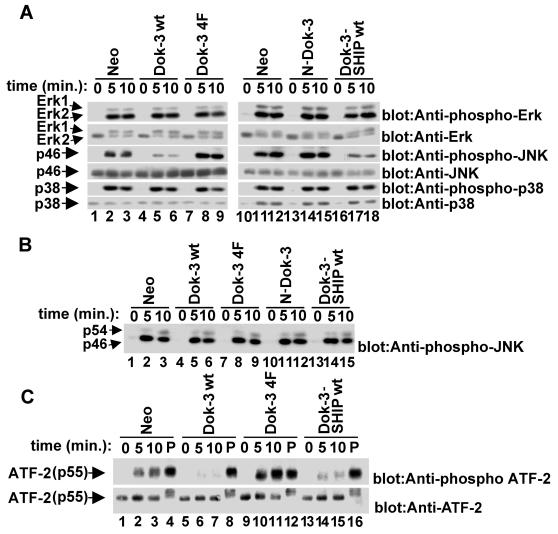
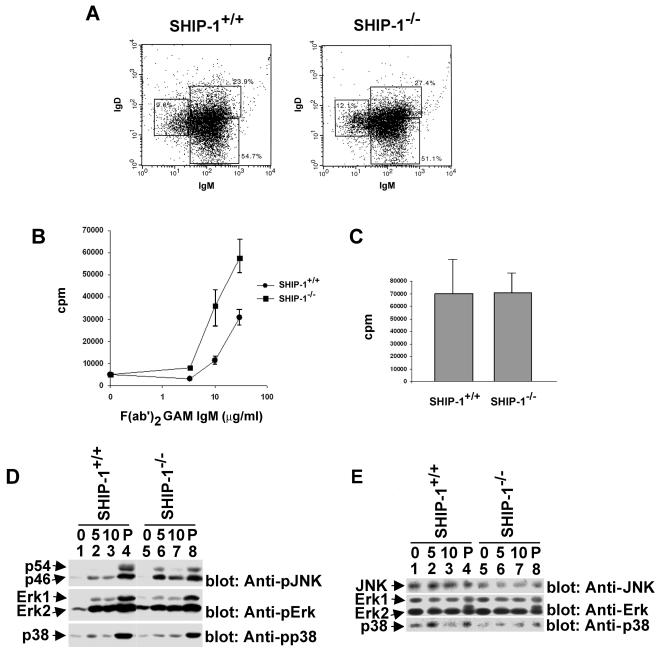
Dok-3 is a Dok-related adaptor expressed in B cells and macrophages. Previously, we reported that Dok-3 is an inhibitor of B-cell activation in A20 B cells and that it associates with SHIP-1, a 5' inositol-specific lipid phosphatase, as well as Csk, a negative regulator of Src kinases. Here, we demonstrate that Dok-3 suppresses B-cell activation by way of its interaction with SHIP-1, rather than Csk. Our biochemical analyses showed that the Dok-3-SHIP-1 complex acts by selectively inhibiting the B-cell receptor (BCR)-evoked activation of the Jun N-terminal protein kinase (JNK) cascade without affecting overall protein tyrosine phosphorylation or activation of previously described SHIP-1 targets like Btk and Akt/PKB. Studies of B cells derived from SHIP-1-deficient mice showed that BCR-triggered activation of JNK is enhanced in the absence of SHIP-1, implying that the Dok-3-SHIP-1 complex (or a related mechanism) is a physiological negative regulator of the JNK cascade in normal B cells. Together, these data elucidate the mechanism by which Dok-3 inhibits B-cell activation. Furthermore, they provide evidence that SHIP-1 can be a negative regulator of JNK signaling in B cells.
Figures
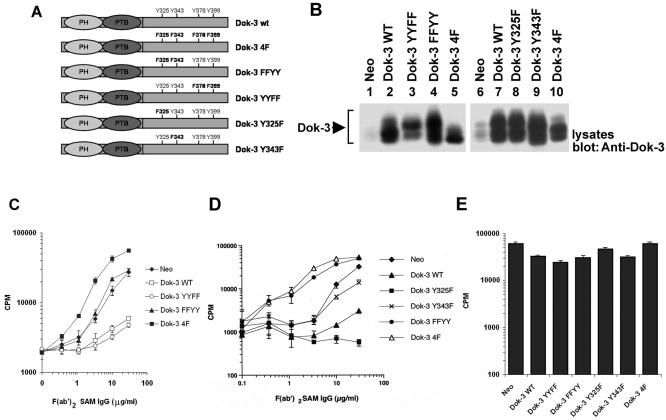
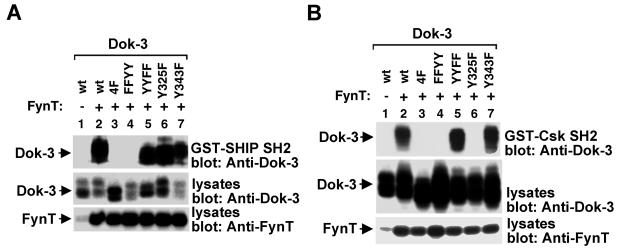
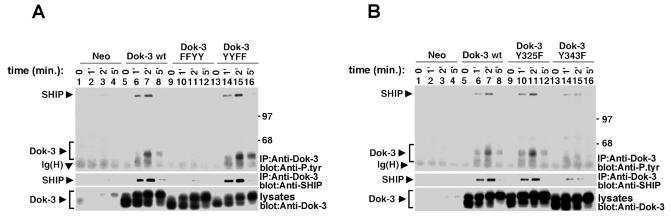
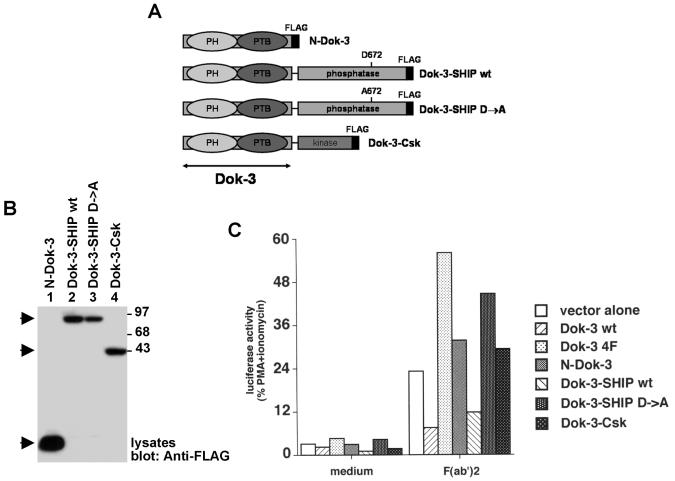
References
-
- Abraham, N., M. C. Miceli, J. R. Parnes, and A. Veillette. 1991. Enhancement of T-cell responsiveness by the lymphocyte-specific tyrosine protein kinase p56lck. Nature 350:62-66. - PubMed
-
- Aman, M. J., T. D. Lamkin, H. Okada, T. Kurosaki, and K. S. Ravichandran. 1998. The inositol phosphatase SHIP inhibits Akt/PKB activation in B cells. J. Biol. Chem. 273:33922-33928. - PubMed
-
- Benschop, R. J., and J. C. Cambier. 1999. B cell development: signal transduction by antigen receptors and their surrogates. Curr. Opin. Immunol. 11:143-151. - PubMed
-
- Bolland, S., R. N. Pearse, T. Kurosaki, and J. V. Ravetch. 1998. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity 8:509-516. - PubMed
-
- Brauweiler, A., I. Tamir, J. Dal Porto, R. J. Benschop, C. D. Helgason, R. K. Humphries, J. H. Freed, and J. C. Cambier. 2000. Differential regulation of B cell development, activation, and death by the src homology 2 domain-containing 5′ inositol phosphatase (SHIP). J. Exp. Med. 191:1545-1554. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous