

A novel retinoic acid-responsive element regulates retinoic acid-induced BLR1 expression
- PMID: 14993281
- PMCID: PMC355834
- DOI: 10.1128/MCB.24.6.2423-2443.2004
A novel retinoic acid-responsive element regulates retinoic acid-induced BLR1 expression
Abstract
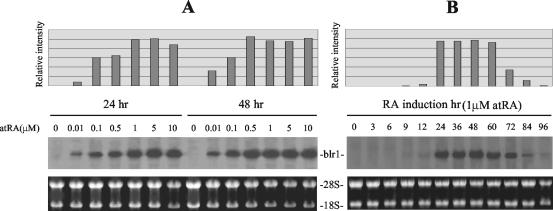
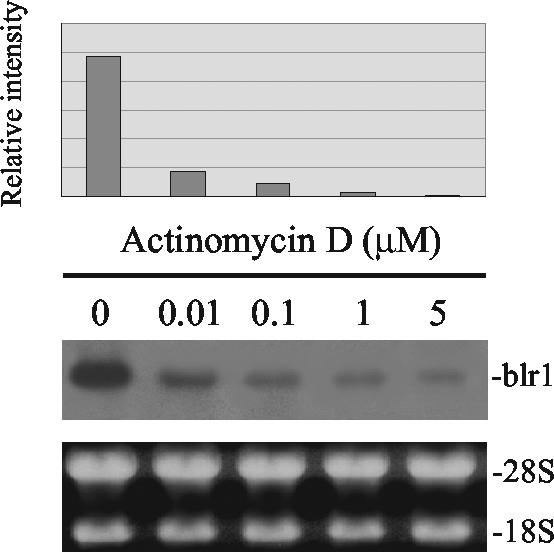
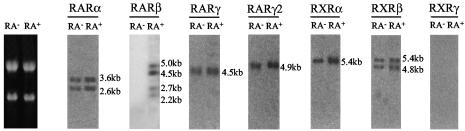
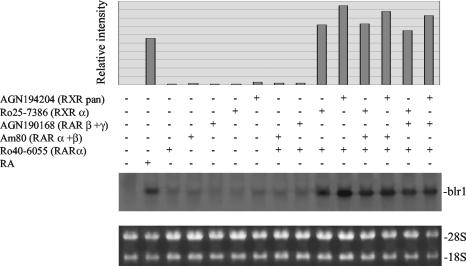
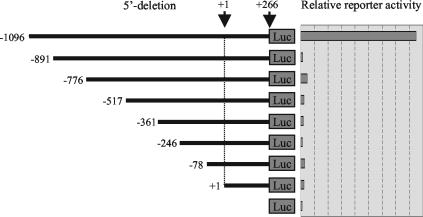
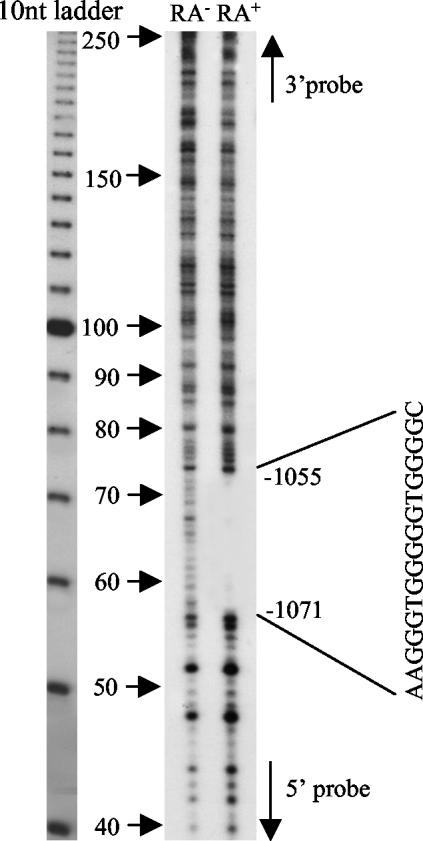
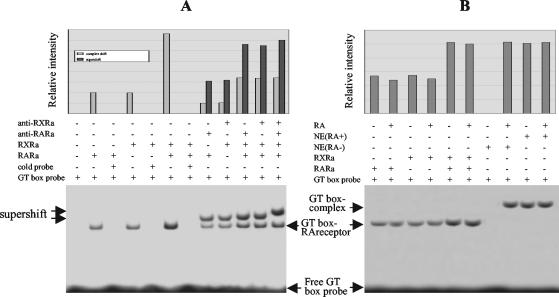
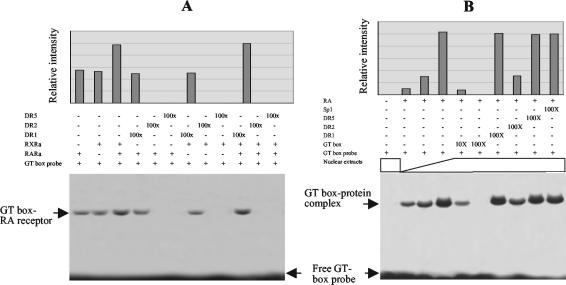
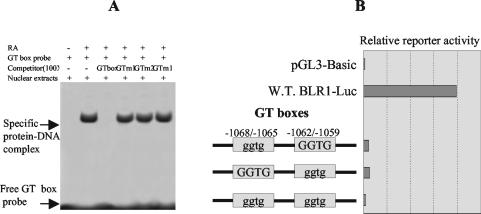
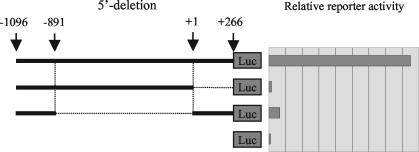
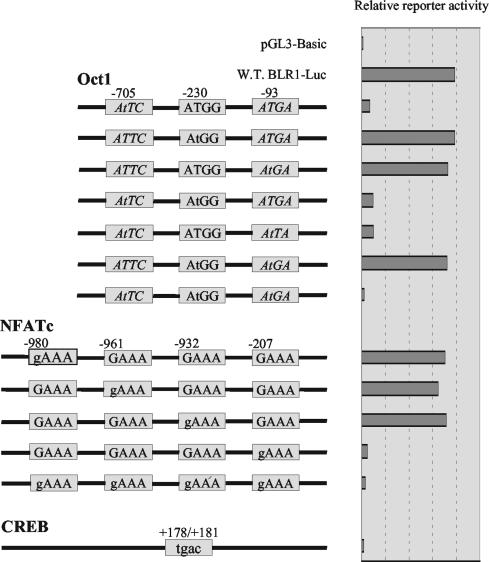
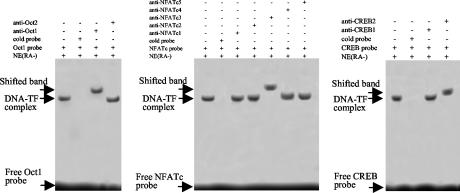
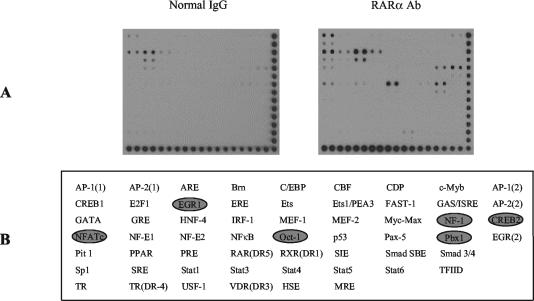
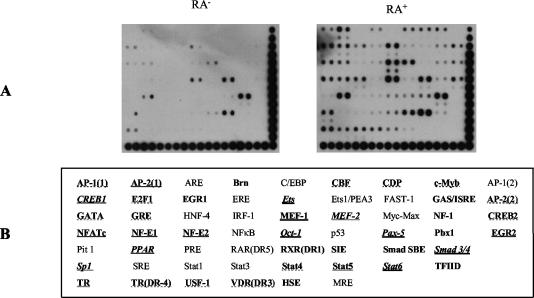
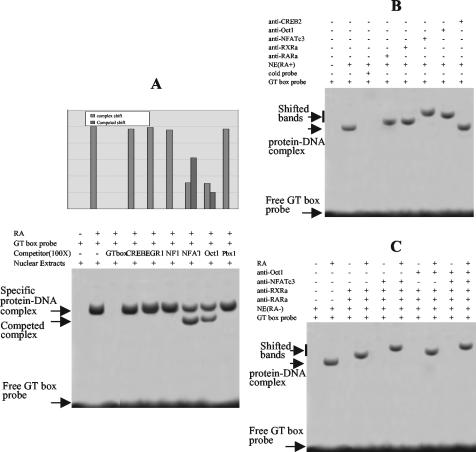
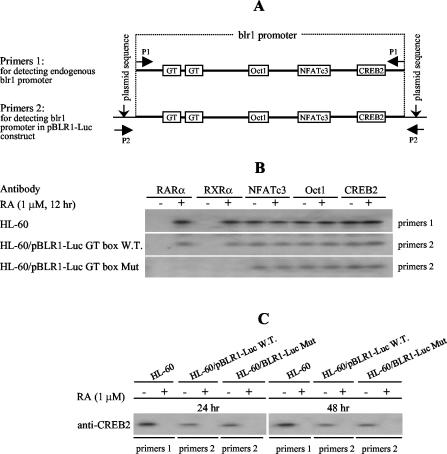
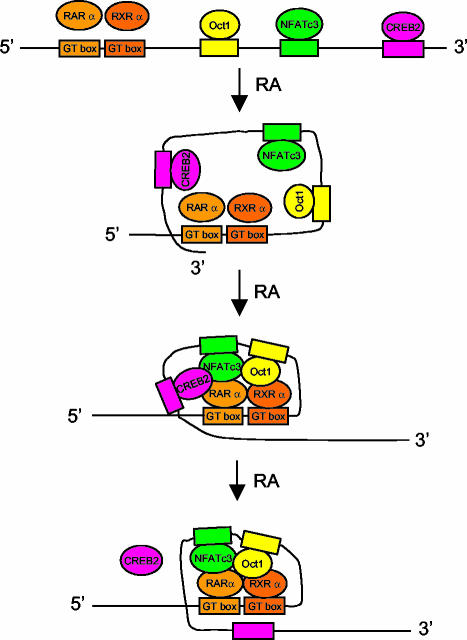
The mechanism of action of retinoic acid (RA) is of broad relevance to cell and developmental biology, nutrition, and cancer chemotherapy. RA is known to induce expression of the Burkitt's lymphoma receptor 1 (BLR1) gene which propels RA-induced cell cycle arrest and differentiation of HL-60 human myeloblastic leukemia cells, motivating the present analysis of transcriptional regulation of blr1 expression by RA. The RA-treated HL-60 cells used here expressed all RA receptor (RAR) and retinoid X receptor (RXR) subtypes (as detected by Northern analysis) except RXRgamma. Treatment with RAR- and RXR-selective ligands showed that RARalpha synergized with RXRalpha to transcriptionally activate blr1 expression. A 5'-flanking region capable of supporting RA-induced blr1 activation in HL-60 cells was found to contain a 205-bp sequence in the distal portion that was necessary for transcriptional activation by RA. Within this sequence DNase I footprinting revealed that RA induced binding of a nuclear protein complex to an element containing two GT boxes. Electromobility shift assays (EMSAs) and supershift assays showed that this element bound recombinant RARalpha and RXRalpha. Without RA there was neither complex binding nor transcriptional activation. Both GT boxes were needed for binding the complex, and mutation of either GT box caused the loss of transcriptional activation by RA. The ability of this cis-acting RAR-RXR binding element to activate transcription in response to RA also depended on downstream sequences where an octamer transcription factor 1 (Oct1) site and a nuclear factor of activated T cells (NFATc) site between this element and the transcriptional start, as well as a cyclic AMP response element binding factor (CREB) site between the transcriptional start and first exon of the blr1 gene, were necessary. Each of these sites bound its corresponding transcription factor. A transcription factor-transcription factor binding array analysis of nuclear lysate from RA-treated cells indicated several prominent RARalpha binding partners; among these, Oct1, NFATc3, and CREB2 were identified by competition EMSA and supershift and chromatin immunoprecipitation assays as components of the complex. RA upregulated expression of these three factors. In sum the results of the present study indicate that RA-induced expression of blr1 expression depends on a novel RA response element. This cis-acting element approximately 1 kb upstream of the transcriptional start consists of two GT boxes that bind RAR and RXR in a nuclear protein complex that also contains Oct1, NFATc3, and CREB2 bound to their cognate downstream consensus binding sites.
Figures

References
-
- Altucci, L., A. Rossin, W. Raffelsberger, A. Reitmair, C. Chomienne, and H. Gronemeyer. 2001. Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat. Med. 7:680-686. - PubMed
-
- Anderson, K. L., K. A. Smith, K. Conners, S. R. McKercher, R. Maki, and B. E. Torbett. 1998. Myeloid development is selectively disrupted in PU. 1 null mice. Blood 91:3702-3710. - PubMed
-
- Babina, M., K. Mammeri, and B. M. Henz. 2001. Retinoic acid up-regulates myeloid ICAM-3 expression and function in a cell-specific fashion—evidence for retinoid signaling pathways in the mast cell lineage. J. Leukoc. Biol. 69:361-371. - PubMed
-
- Battle, T. E., and A. Yen. 2002. Ectopic expression of CXCR5/BLR1 accelerates retinoic acid- and vitamin D3-induced monocytic differentiation of U937 cells. Exp. Biol. Med. 227:753-762. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous