

Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element
- PMID: 14993291
- PMCID: PMC355825
- DOI: 10.1128/MCB.24.6.2546-2559.2004
Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element
Abstract
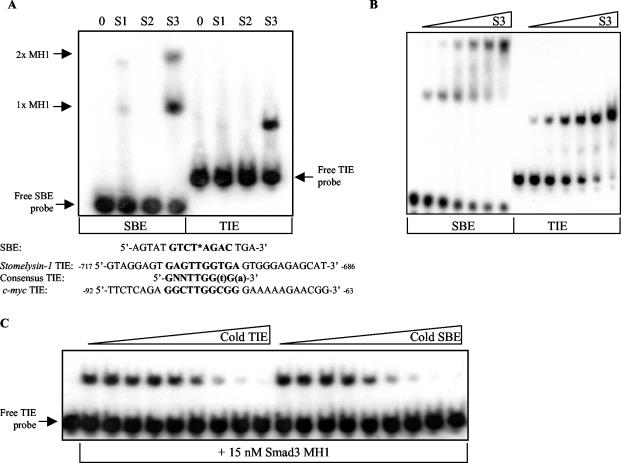
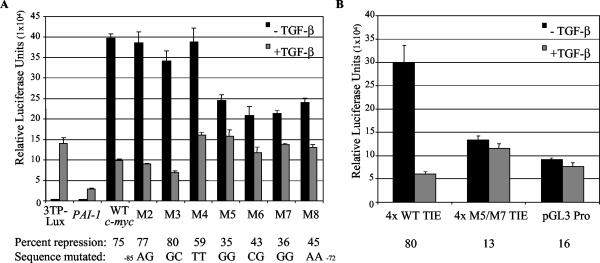
Smad proteins are the most well-characterized intracellular effectors of the transforming growth factor beta (TGF-beta) signal. The ability of the Smads to act as transcriptional activators via TGF-beta-induced recruitment to Smad binding elements (SBE) within the promoters of TGF-beta target genes has been firmly established. However, the elucidation of the molecular mechanisms involved in TGF-beta-mediated transcriptional repression are only recently being uncovered. The proto-oncogene c-myc is repressed by TGF-beta, and this repression is required for the manifestation of the TGF-beta cytostatic program in specific cell types. We have shown that Smad3 is required for both TGF-beta-induced repression of c-myc and subsequent growth arrest in keratinocytes. The transcriptional repression of c-myc is dependent on direct Smad3 binding to a novel Smad binding site, termed a repressive Smad binding element (RSBE), within the TGF-beta inhibitory element (TIE) of the c-myc promoter. The c-myc TIE is a composite element, comprised of an overlapping RSBE and a consensus E2F site, that is capable of binding at least Smad3, Smad4, E2F-4, and p107. The RSBE is distinct from the previously defined SBE and may partially dictate, in conjunction with the promoter context of the overlapping E2F site, whether the Smad3-containing complex actively represses, as opposed to transactivates, the c-myc promoter.
Figures





Similar articles
-
c-myc is a downstream target of the Smad pathway.J Biol Chem. 2002 Jan 4;277(1):854-61. doi: 10.1074/jbc.M104170200. Epub 2001 Oct 31. J Biol Chem. 2002. PMID: 11689553
-
E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression.Cell. 2002 Jul 12;110(1):19-32. doi: 10.1016/s0092-8674(02)00801-2. Cell. 2002. PMID: 12150994
-
E2F4-RB and E2F4-p107 complexes suppress gene expression by transforming growth factor beta through E2F binding sites.Proc Natl Acad Sci U S A. 1997 May 13;94(10):4948-53. doi: 10.1073/pnas.94.10.4948. Proc Natl Acad Sci U S A. 1997. PMID: 9144170 Free PMC article.
-
Structural and functional characterization of the transforming growth factor-beta -induced Smad3/c-Jun transcriptional cooperativity.J Biol Chem. 2000 Dec 8;275(49):38802-12. doi: 10.1074/jbc.M004731200. J Biol Chem. 2000. PMID: 10995748
-
Nuclear factor YY1 inhibits transforming growth factor beta- and bone morphogenetic protein-induced cell differentiation.Mol Cell Biol. 2003 Jul;23(13):4494-510. doi: 10.1128/MCB.23.13.4494-4510.2003. Mol Cell Biol. 2003. PMID: 12808092 Free PMC article.
Cited by
-
Deptor enhances triple-negative breast cancer metastasis and chemoresistance through coupling to survivin expression.Neoplasia. 2015 Mar;17(3):317-28. doi: 10.1016/j.neo.2015.02.003. Neoplasia. 2015. PMID: 25810016 Free PMC article.
-
Gene expression of the lysophosphatidic acid receptor 1 is a target of transforming growth factor beta.Oncogene. 2013 Jun 27;32(26):3198-206. doi: 10.1038/onc.2012.325. Epub 2012 Jul 23. Oncogene. 2013. PMID: 22824789 Free PMC article.
-
Transcription profiling of lung adenocarcinomas of c-myc-transgenic mice: identification of the c-myc regulatory gene network.BMC Syst Biol. 2008 May 22;2:46. doi: 10.1186/1752-0509-2-46. BMC Syst Biol. 2008. PMID: 18498649 Free PMC article.
-
The role of transforming growth factor β in T helper 17 differentiation.Immunology. 2018 Sep;155(1):24-35. doi: 10.1111/imm.12938. Epub 2018 May 8. Immunology. 2018. PMID: 29682722 Free PMC article. Review.
-
MYC Deregulation in Primary Human Cancers.Genes (Basel). 2017 May 25;8(6):151. doi: 10.3390/genes8060151. Genes (Basel). 2017. PMID: 28587062 Free PMC article. Review.
References
-
- Abdollah, S., M. Macias-Silva, T. Tsukazaki, H. Hayashi, L. Attisano, and J. L. Wrana. 1997. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 272:27678-27685. - PubMed
-
- Akiyoshi, S., H. Inoue, J. Hanai, K. Kusanagi, N. Nemoto, K. Miyazono, and M. Kawabata. 1999. c-Ski acts as a transcriptional co-repressor in transforming growth factor-beta signaling through interaction with smads. J. Biol. Chem. 274:35269-35277. - PubMed
-
- Attisano, L., and J. L. Wrana. 2000. Smads as transcriptional co-modulators. Curr. Opin. Cell Biol. 12:235-243. - PubMed
-
- Baker, J. C., and R. M. Harland. 1997. From receptor to nucleus: the Smad pathway. Curr. Opin. Genet. Dev. 7:467-473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous