

Pharmacogenomics of cystic fibrosis
- PMID: 14993338
- PMCID: PMC8364423
Pharmacogenomics of cystic fibrosis
Abstract
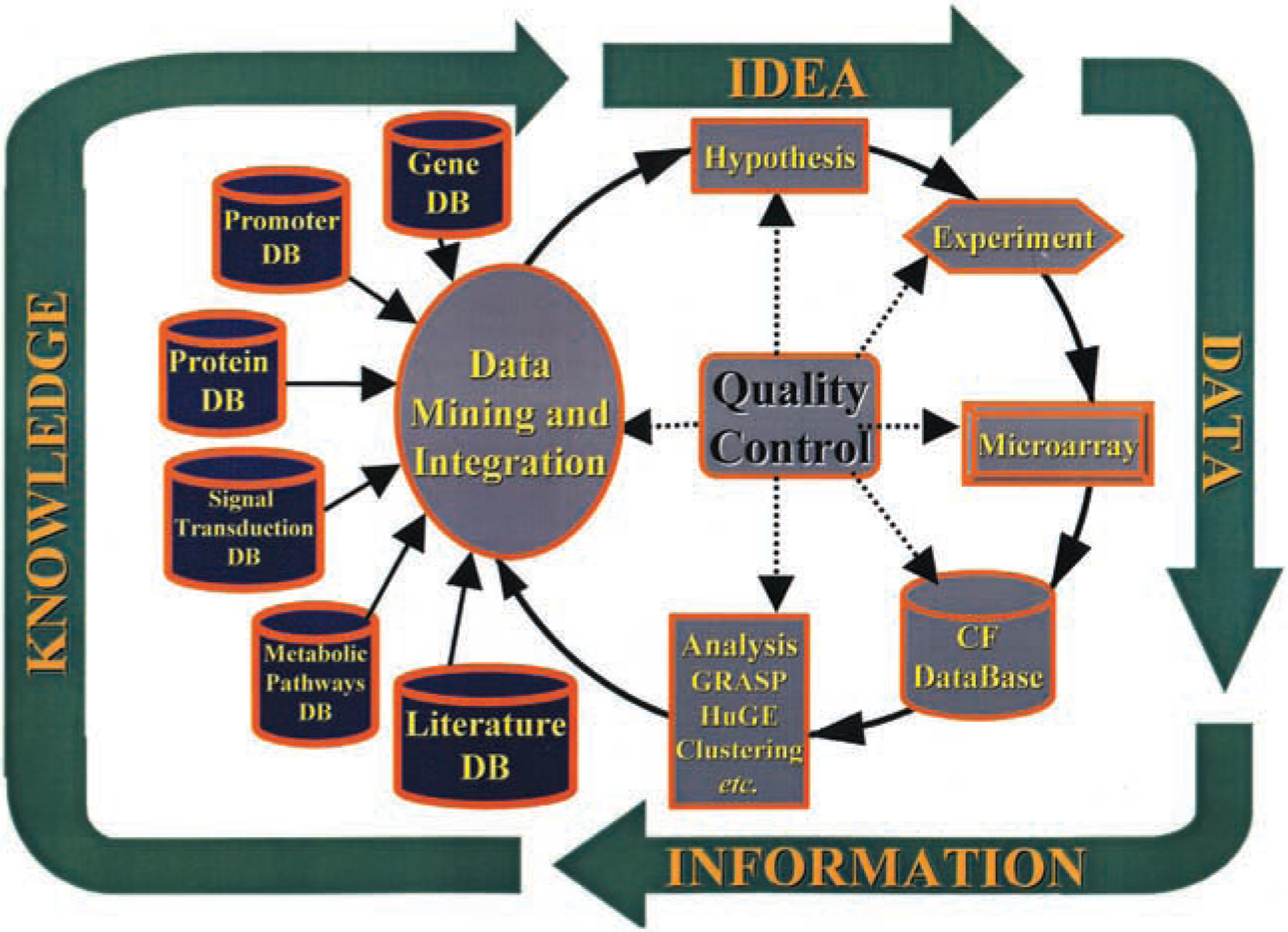
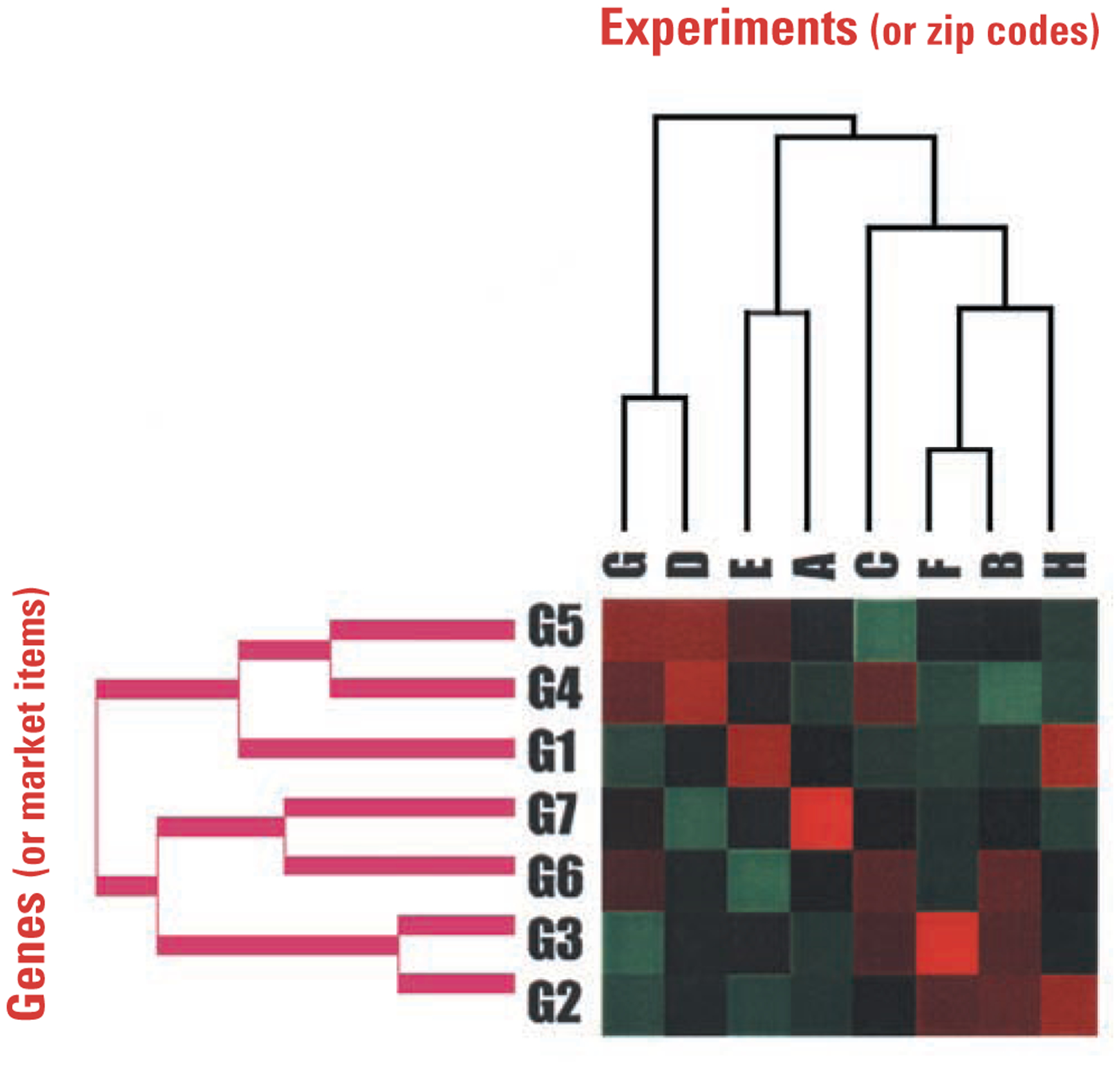
Pharmacogenomics is becoming a frontline instrument of drug discovery, where the drug-dependent patterns of global gene expression are employed as biologically relevant end points. In the case of cystic fibrosis (CF), cells and tissues from CF patients provide the starting points of genomic analysis. The end points for drug discovery are proposed to reside in gene expression patterns of CF cells that have been corrected by gene therapy. A case is made here that successful drug therapy and gene therapy should, hypothetically, converge at a common end point. In response to a virtual tidal wave of genomic data, bioinformatics algorithms are needed to identify those genes that truly reveal drug efficacy. As examples, we describe the hierarchical clustering, GRASP, and GENESAVER algorithms, particularly within a hypothesis-driven context that focuses on data for a CF candidate drug. Pharmacogenomic approaches to CF, and other similar diseases, may eventually give us the opportunity to create drugs that work in a patient- or mutation-specific manner.
Figures



Similar articles
-
Control of the proinflammatory state in cystic fibrosis lung epithelial cells by genes from the TNF-alphaR/NFkappaB pathway.Mol Med. 2001 Aug;7(8):523-34. Mol Med. 2001. PMID: 11591888 Free PMC article.
-
Cystic fibrosis and the use of pharmacogenomics to determine surrogate endpoints for drug discovery.Am J Pharmacogenomics. 2001;1(3):223-38. doi: 10.2165/00129785-200101030-00006. Am J Pharmacogenomics. 2001. PMID: 12083969 Review.
-
Pharmacogenetics of cystic fibrosis treatment.Pharmacogenomics. 2016 Aug;17(13):1453-63. doi: 10.2217/pgs.16.25. Epub 2016 Aug 4. Pharmacogenomics. 2016. PMID: 27490265 Review.
-
Opportunity for pharmacogenomic testing in patients with cystic fibrosis.Pediatr Pulmonol. 2022 Apr;57(4):903-907. doi: 10.1002/ppul.25809. Epub 2022 Jan 11. Pediatr Pulmonol. 2022. PMID: 34967155
-
Towards the pharmacogenomics of cystic fibrosis.Pharmacogenomics. 2002 Jan;3(1):75-87. doi: 10.1517/14622416.3.1.75. Pharmacogenomics. 2002. PMID: 11966405 Review.
Cited by
-
CFTR activity and mitochondrial function.Redox Biol. 2013 Feb 5;1(1):190-202. doi: 10.1016/j.redox.2012.11.007. Redox Biol. 2013. PMID: 24024153 Free PMC article. Review.
-
The Pharmacokinetic Changes in Cystic Fibrosis Patients Population: Narrative Review.Medicines (Basel). 2024 Dec 31;12(1):1. doi: 10.3390/medicines12010001. Medicines (Basel). 2024. PMID: 39846711 Free PMC article. Review.
References
-
- Lockhart DJ and Winzeler DA Genomics, gene expression and DNA arrays. Nature 405, 827–836 (2000). - PubMed
-
- Debouck C and Metcalf B The impact of genomics on drug discovery. Annu.Rev. Pharmacol. Toxicol 40, 193–207 (2000). - PubMed
-
- Young RA Biomedical discovery with DNA arrays. Cell 102, 9–15 (2000). - PubMed
-
- Roses AD Pharmacogenetics and the practice of medicine. Nature 405, 857–865 (2000). - PubMed
-
- Pollard HB Anatomical Genomics: Systems of genes underlying the biology of systems. Anatomical Record 259, iii–ix (2000). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials