

Targeting oestrogen to kill the cancer but not the patient
- PMID: 14997187
- PMCID: PMC2409614
- DOI: 10.1038/sj.bjc.6601627
Targeting oestrogen to kill the cancer but not the patient
Abstract
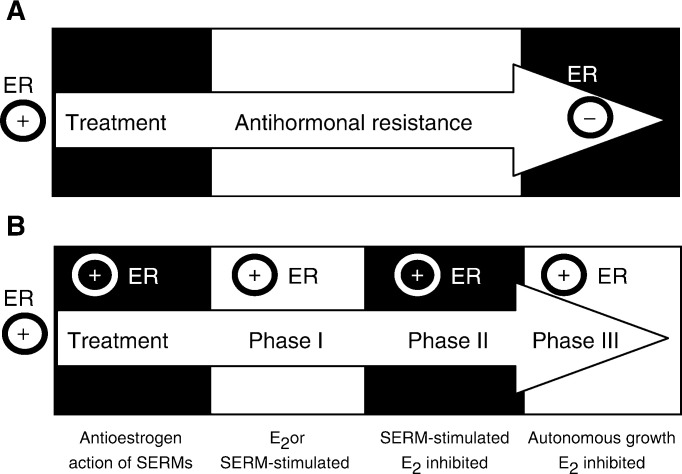
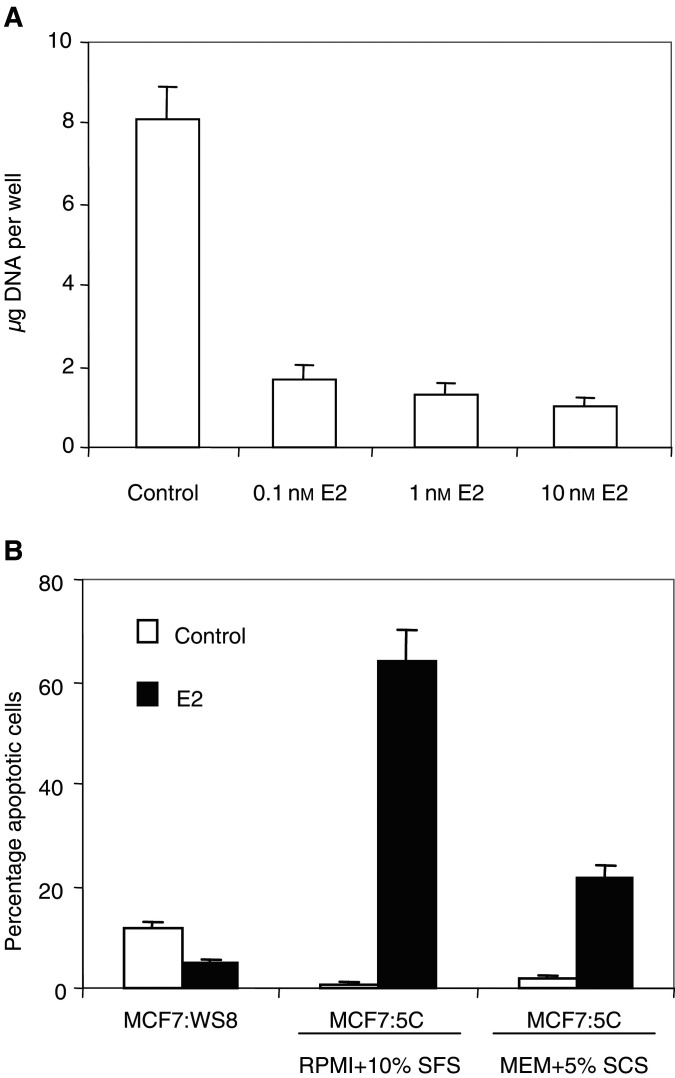
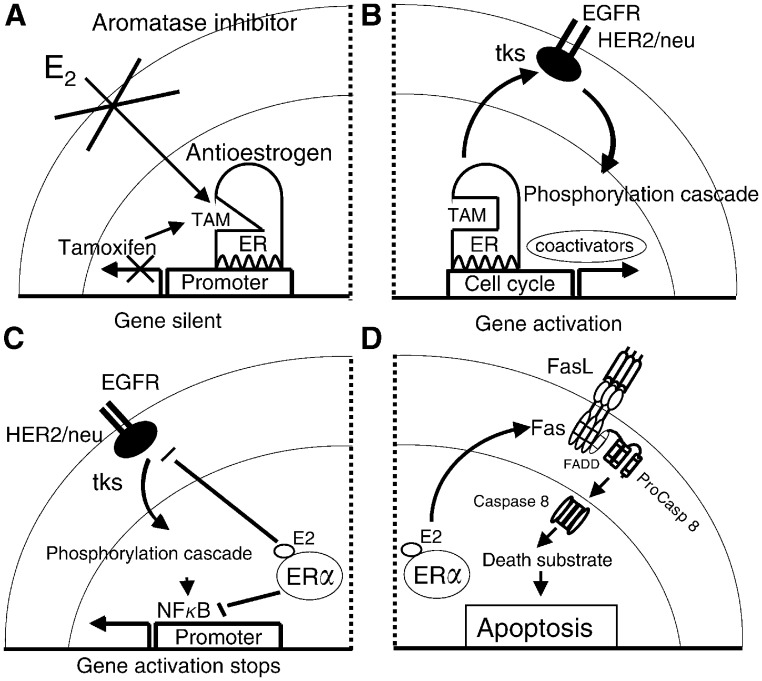
The link between sex steroids and the development and growth of breast cancer has proved to be an invaluable clue for advances in the prevention and treatment of breast cancer. The identification of the oestrogen receptor (ER) not only allowed advances in the molecular endocrinology of oestrogen action, but also provided a target for antioestrogenic therapeutic agents. However, the application of long-term or indefinite treatment regimens has consequences for the breast cancer. New forms of resistance, based upon enhanced cellular survival networks independent of ER and the suppression of apoptotic mechanisms, develop and then evolve. Remarkably, low concentrations of oestrogen collapse survival pathways and induce apoptosis in completely antihormonally refractory breast cancer. However, recurrent oestrogen-stimulated disease is again sensitive to antihormonal therapy. The novel reapplication of the ER as a therapeutic target for apoptosis is emerging as a new strategy for the long-term targeted maintenance treatment of breast cancer, and in formulating a targeted strategy for endocrine independent cancer.
Figures
References
-
- ATAC Trialists' Group (2002) Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomized trial. Lancet 359: 2131–2139 - PubMed
-
- Bentrem D, Fox JE, Pearce ST, Liu H, Pappas S, Kupfer D, Zapf JW, Jordan VC (2003) Distinct moleular conformations of the estrogen receptor alpha complex exploited by environmental estrogens. Cancer Res 63: 7490–7496 - PubMed
-
- Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, Norton L, Nickelsen T, Bjarnason NH, Morrow M, Lippman ME, Black D, Glusman JE, Costa A, Jordan VC (1999) The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 281: 2189–2197 - PubMed
-
- Cuzick J, Powles T, Veronesi U, Forbes J, Edwards R, Ashley S, Boyle P (2003) Overview of the main outcomes in breast-cancer prevention trials. Lancet 361: 296–300 - PubMed
-
- EBCTCG (1998) Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 351: 1451–1467 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
