

Positive selection detection in 40,000 human immunodeficiency virus (HIV) type 1 sequences automatically identifies drug resistance and positive fitness mutations in HIV protease and reverse transcriptase
- PMID: 15016892
- PMCID: PMC371046
- DOI: 10.1128/jvi.78.7.3722-3732.2004
Positive selection detection in 40,000 human immunodeficiency virus (HIV) type 1 sequences automatically identifies drug resistance and positive fitness mutations in HIV protease and reverse transcriptase
Abstract
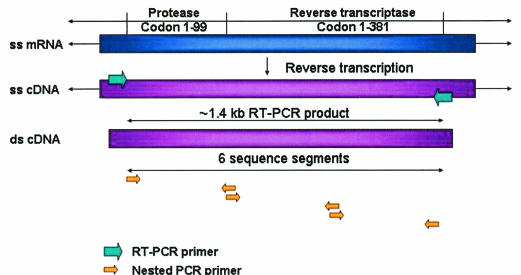
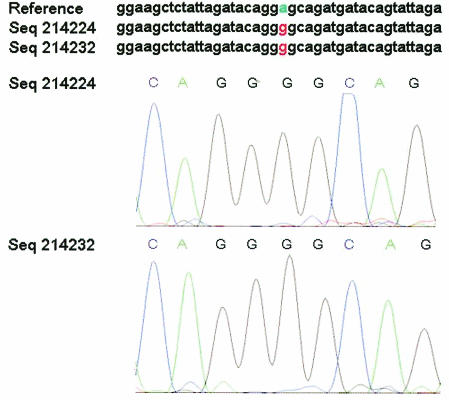
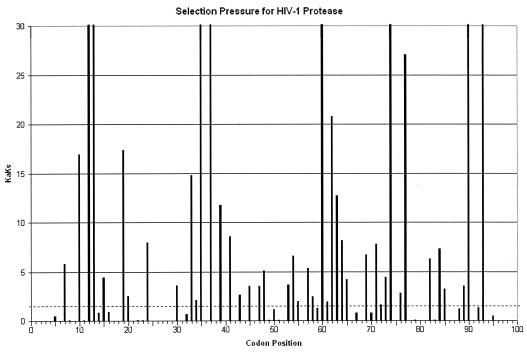
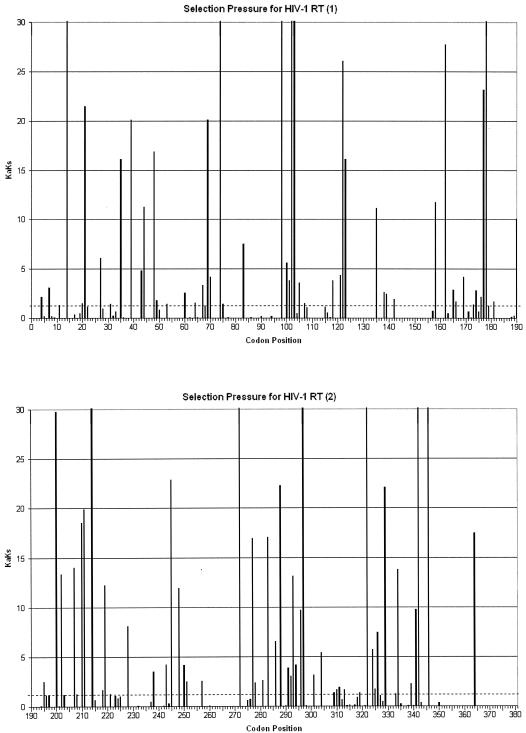
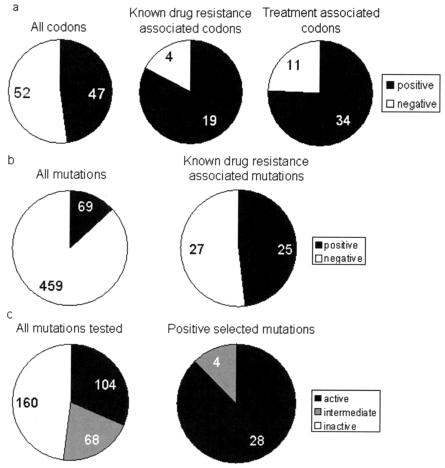
Drug resistance is a major problem in the treatment of AIDS, due to the very high mutation rate of human immunodeficiency virus (HIV) and subsequent rapid development of resistance to new drugs. Identification of mutations associated with drug resistance is critical for both individualized treatment selection and new drug design. We have performed an automated mutation analysis of HIV Type 1 (HIV-1) protease and reverse transcriptase (RT) from approximately 40,000 AIDS patient plasma samples sequenced by Specialty Laboratories Inc. from 1999 to mid-2002. This data set provides a nearly complete mutagenesis of HIV protease and enables the calculation of statistically significant K(a)/K(s) values for each individual amino acid mutation in protease and RT. Positive selection (i.e., a K(a)/K(s) ratio of >1, indicating increased reproductive fitness) detected 19 of 23 known drug-resistant mutation positions in protease and 20 of 34 such positions in RT. We also discovered 163 new amino acid mutations in HIV protease and RT that are strong candidates for drug resistance or fitness. Our results match available independent data on protease mutations associated with specific drug treatments and mutations with positive reproductive fitness, with high statistical significance (the P values for the observed matches to occur by random chance are 10(-5.2) and 10(-16.6), respectively). Our mutation analysis provides a valuable resource for AIDS research and will be available to academic researchers upon publication at http://www.bioinformatics.ucla.edu/HIV. Our data indicate that positive selection mapping is an analysis that can yield powerful insights from high-throughput sequencing of rapidly mutating pathogens.
Figures
References
-
- Bally, F., R. Martinez, S. Peters, P. Sudre, and A. Telenti. 2000. Polymorphism of HIV type 1 gag p7/p1 and p1/p6 cleavage sites: clinical significance and implications for resistance to protease inhibitors. AIDS Res. Hum. Retrovir. 16:1209-1213. - PubMed
-
- Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483-489. - PubMed
-
- Ewing, B., L. Hillier, M. C. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175-185. - PubMed
-
- Hu, G., B. Modrek, H. M. F. R. Stensland, J. Saarela, P. Pajukanta, V. Kustanovich, L. Peltonen, S. F. Nelson, and C. Lee. 2002. Efficient discovery of single-nucleotide polymorphisms in coding regions of human genes. Pharmacogenomics J. 2:236-242. - PubMed
