

Metagenes and molecular pattern discovery using matrix factorization
- PMID: 15016911
- PMCID: PMC384712
- DOI: 10.1073/pnas.0308531101
Metagenes and molecular pattern discovery using matrix factorization
Abstract
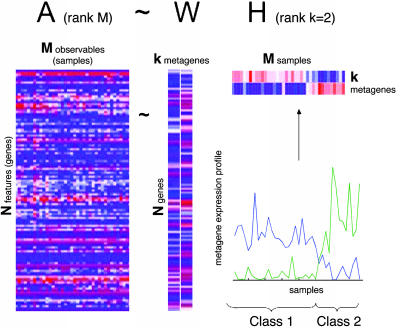
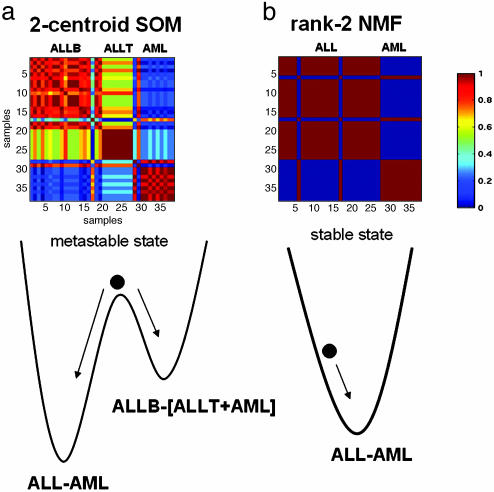
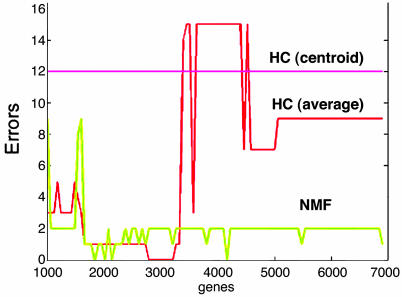
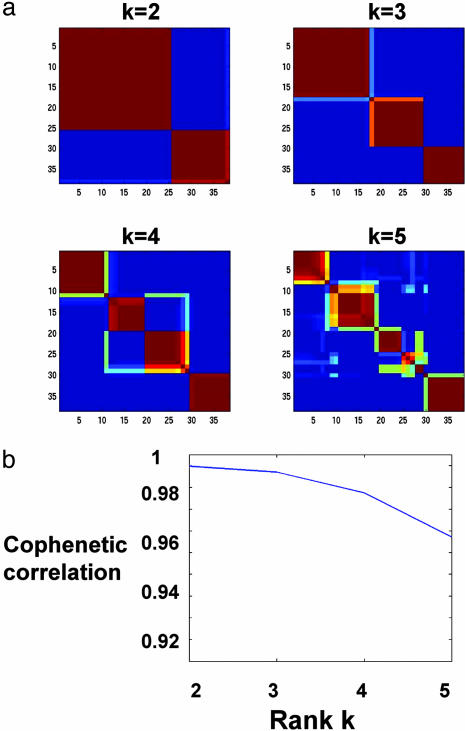
We describe here the use of nonnegative matrix factorization (NMF), an algorithm based on decomposition by parts that can reduce the dimension of expression data from thousands of genes to a handful of metagenes. Coupled with a model selection mechanism, adapted to work for any stochastic clustering algorithm, NMF is an efficient method for identification of distinct molecular patterns and provides a powerful method for class discovery. We demonstrate the ability of NMF to recover meaningful biological information from cancer-related microarray data. NMF appears to have advantages over other methods such as hierarchical clustering or self-organizing maps. We found it less sensitive to a priori selection of genes or initial conditions and able to detect alternative or context-dependent patterns of gene expression in complex biological systems. This ability, similar to semantic polysemy in text, provides a general method for robust molecular pattern discovery.
Figures
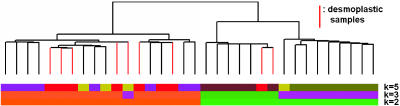
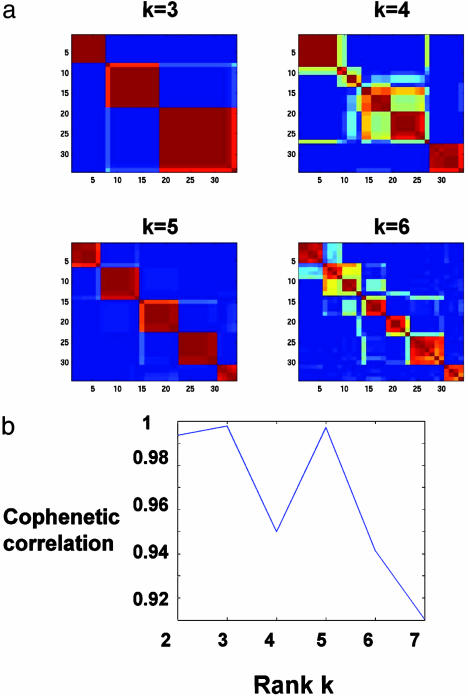
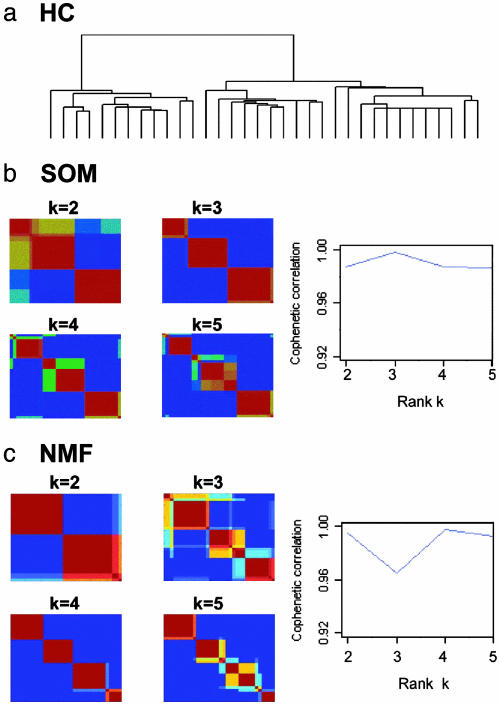
References
-
- Alizadeh, A. A., Eisen, M. B., Davis, R. E., Ma, C., Lossos, I. S., Rosenwald, A., Boldrick, J. C., Sabet, H., Tran, T., Yu, X., et al. (2000) Nature 403, 503–511. - PubMed
-
- Perou, C. M., Sorlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., Pollack, J. R., Ross, D. T., Johnsen, H., Akslen, L. A., et al. (2000) Nature 406, 747–752. - PubMed
-
- Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J. P., Coller, H., Loh, M. L., Downing, J. R., Caligiuri, M. A., et al. (1999) Science 286, 531–537. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
