

S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism
- PMID: 15024124
- PMCID: PMC384724
- DOI: 10.1073/pnas.0400658101
S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism
Abstract
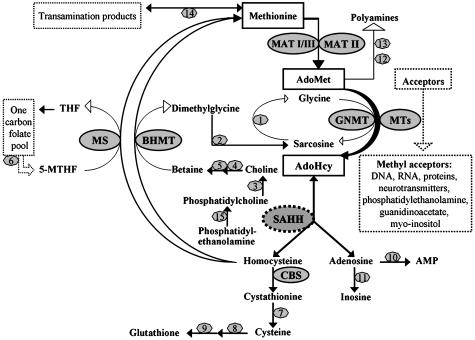
We report studies of a Croatian boy, a proven case of human S-adenosylhomocysteine (AdoHcy) hydrolase deficiency. Psychomotor development was slow until his fifth month; thereafter, virtually absent until treatment was started. He had marked hypotonia with elevated serum creatine kinase and transaminases, prolonged prothrombin time and low albumin. Electron microscopy of muscle showed numerous abnormal myelin figures; liver biopsy showed mild hepatitis with sparse rough endoplasmic reticulum. Brain MRI at 12.7 months revealed white matter atrophy and abnormally slow myelination. Hypermethioninemia was present in the initial metabolic study at age 8 months, and persisted (up to 784 microM) without tyrosine elevation. Plasma total homocysteine was very slightly elevated for an infant to 14.5-15.9 microM. In plasma, S-adenosylmethionine was 30-fold and AdoHcy 150-fold elevated. Activity of AdoHcy hydrolase was approximately equal to 3% of control in liver and was 5-10% of the control values in red blood cells and cultured fibroblasts. We found no evidence of a soluble inhibitor of the enzyme in extracts of the patient's cultured fibroblasts. Additional pretreatment abnormalities in plasma included low concentrations of phosphatidylcholine and choline, with elevations of guanidinoacetate, betaine, dimethylglycine, and cystathionine. Leukocyte DNA was hypermethylated. Gene analysis revealed two mutations in exon 4: a maternally derived stop codon, and a paternally derived missense mutation. We discuss reasons for biochemical abnormalities and pathophysiological aspects of AdoHcy hydrolase deficiency.
Figures


References
-
- De La Haba, G. & Cantoni, G. L. (1959) J. Biol. Chem. 234, 603-608. - PubMed
-
- Cantoni, G. L. & Chiang, P. K. (1980) in Novel Biochemical and Structural Aspects, eds. Cavallini, D., Gaull, G. E. & Zappia, V. (Plenum, New York), pp. 67-80.
-
- Clarke, S. & Banfield, K. (2001) in Homocysteine in Health and Disease, eds. Carmel, R. & Jacobsen, D. W. (Cambridge Univ. Press, Cambridge, U.K.), pp. 63-78.
-
- Hoffman, D. R., Cornatzer, W. E. & Duerre, J. A. (1979) Can. J. Biochem. 57, 56-65. - PubMed
-
- Allen, R. H., Stabler, S. P. & Lindenbaum, J. (1993) Metabolism 42, 1448-1460. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
