

Database analyses for the prediction of in vivo drug-drug interactions from in vitro data
- PMID: 15025746
- PMCID: PMC1884485
- DOI: 10.1111/j.1365-2125.2003.02041.x
Database analyses for the prediction of in vivo drug-drug interactions from in vitro data
Erratum in
- Br J Clin Pharmacol. 2004 Nov;58(5):565-8
Abstract
Aims: In theory, the magnitude of an in vivo drug-drug interaction arising from the inhibition of metabolic clearance can be predicted using the ratio of inhibitor concentration ([I]) to inhibition constant (K(i)). The aim of this study was to construct a database for the prediction of drug-drug interactions from in vitro data and to evaluate the use of the various estimates for the inhibitor concentrations in the term [I]/K(i).
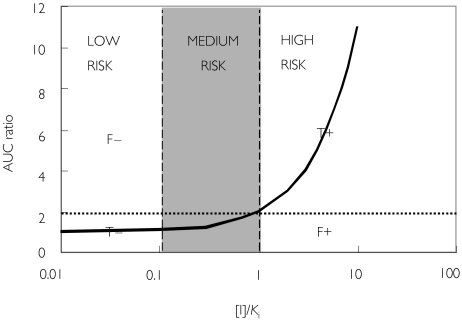
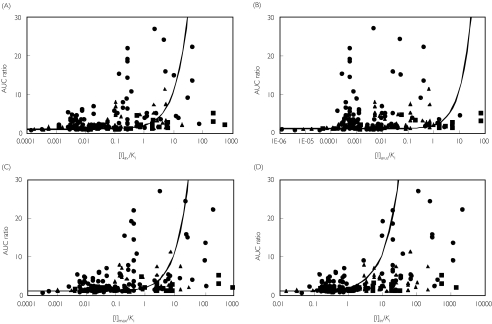
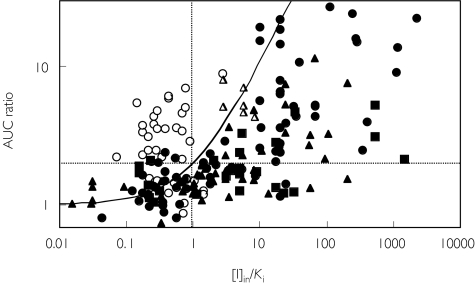
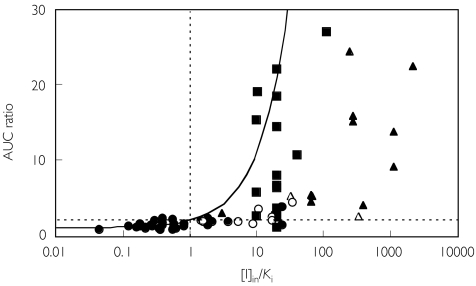
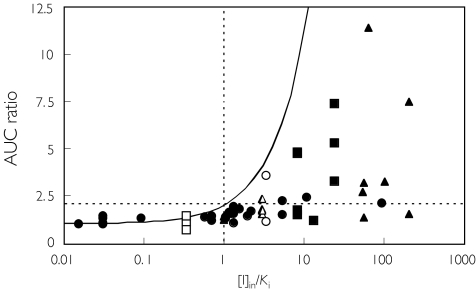
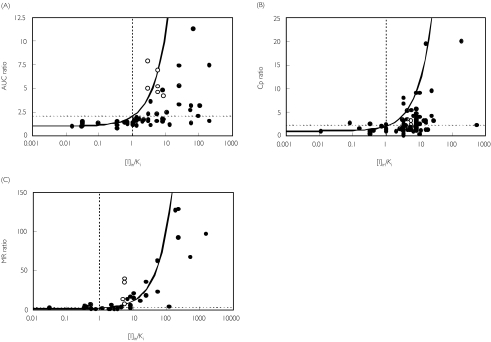
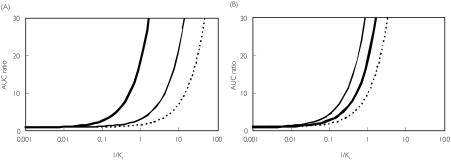
Methods: One hundred and ninety-three in vivo drug-drug interaction studies involving inhibition of CYP3A4, CYP2D6 or CYP2C9 were collated from the literature together with in vitro K(i) values and pharmacokinetic parameters for inhibitors, to allow calculation of average/maximum systemic plasma concentration during the dosing interval and maximum hepatic input plasma concentration (both total and unbound concentration). The observed increase in AUC (decreased clearance) was plotted against the estimated [I]/K(i) ratio for qualitative zoning of the predictions.
Results: The incidence of false negative predictions (AUC ratio > 2, [I]/K(i) < 1) was largest using the average unbound plasma concentration and smallest using the hepatic input total plasma concentration of inhibitor for each of the CYP enzymes. Excluding mechanism-based inhibition, the use of total hepatic input concentration resulted in essentially no false negative predictions, though several false positive predictions (AUC ratio < 2, [I]/K(i) > 1) were found. The incidence of true positive predictions (AUC ratio > 2, [I]/K(i) > 1) was also highest using the total hepatic input concentration.
Conclusions: The use of the total hepatic input concentration of inhibitor together with in vitro K(i) values was the most successful method for the categorization of putative CYP inhibitors and for identifying negative drug-drug interactions. However this approach should be considered as an initial discriminating screen, as it is empirical and requires subsequent mechanistic studies to provide a comprehensive evaluation of a positive result.
Figures
Comment in
-
Drug interactions-information, education, and the British National Formulary.Br J Clin Pharmacol. 2004 Apr;57(4):371-2. doi: 10.1111/j.1365-2125.2004.02125.x. Br J Clin Pharmacol. 2004. PMID: 15025733 Free PMC article. No abstract available.
References
-
- Segel IH. Enzyme KineticsBehaviour and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. New York: Wiley & Sons Inc; 1975.
-
- Zomorodi K, Houston JB. Effect of omeprazole on diazepam disposition in the rat: in vitro and in vivo studies. Pharm Res. 1995;12:1642–6. - PubMed
-
- Davis JD, Aarons L, Houston JB. Relationship between enoxacin and ciprofloxacin plasma concentrations and theophylline disposition. Pharm Res. 1994;11:1424–8. - PubMed
-
- Ervine CM, Matthew DE, Brennan B, Jezequel SG, Humphrey MJ, Houston JB. Comparison of the steady-state pharmacokinetics of fluconazole and ketoconazole and their relative effects on cytochrome P450 activity in rats: use of antipyrine and a steady-state infusion approach to assess plasma concentration-response relationships. Drug Metab Dispos. 1996;24:211–5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
