

Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage
- PMID: 15044535
- PMCID: PMC6729853
- DOI: 10.1523/JNEUROSCI.0155-04.2004
Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage
Abstract
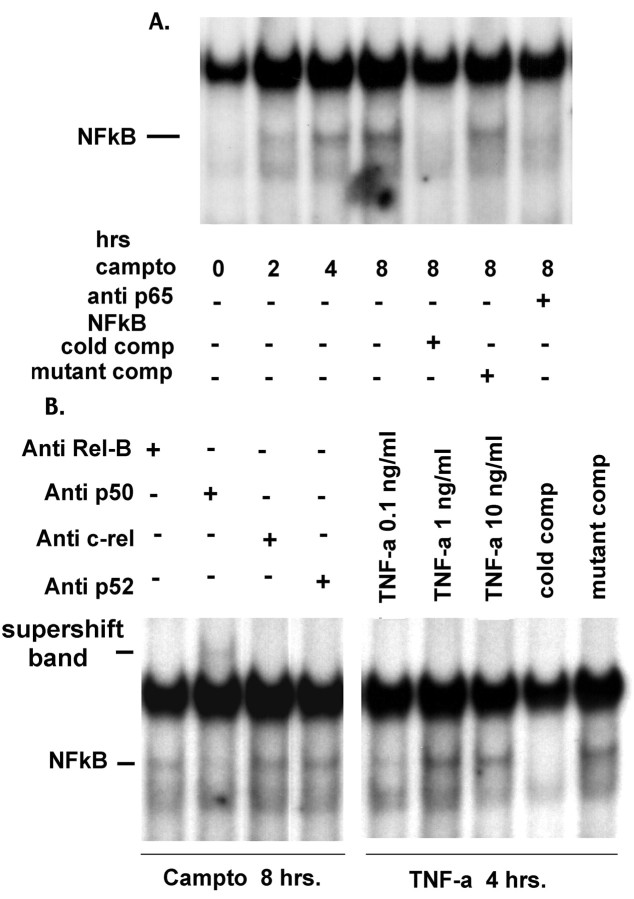
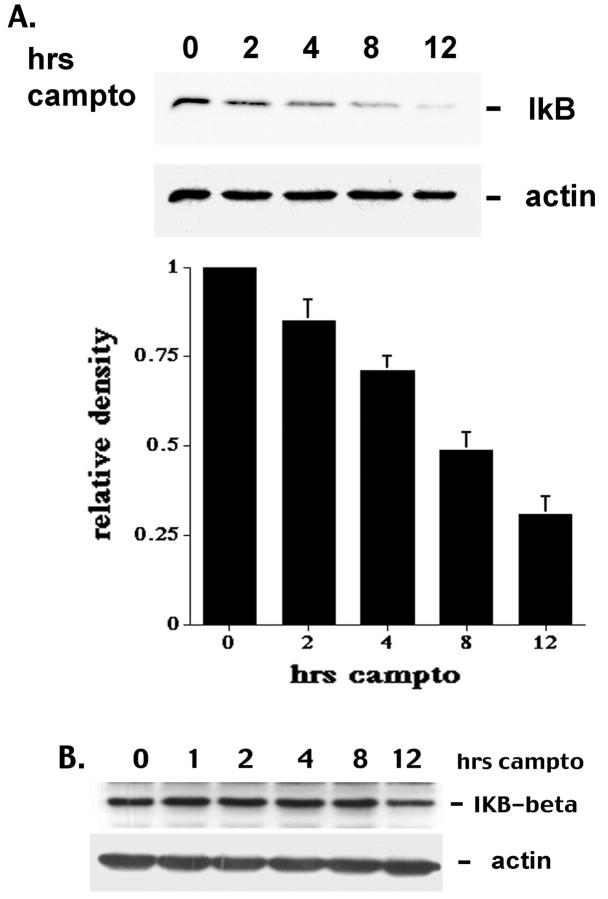
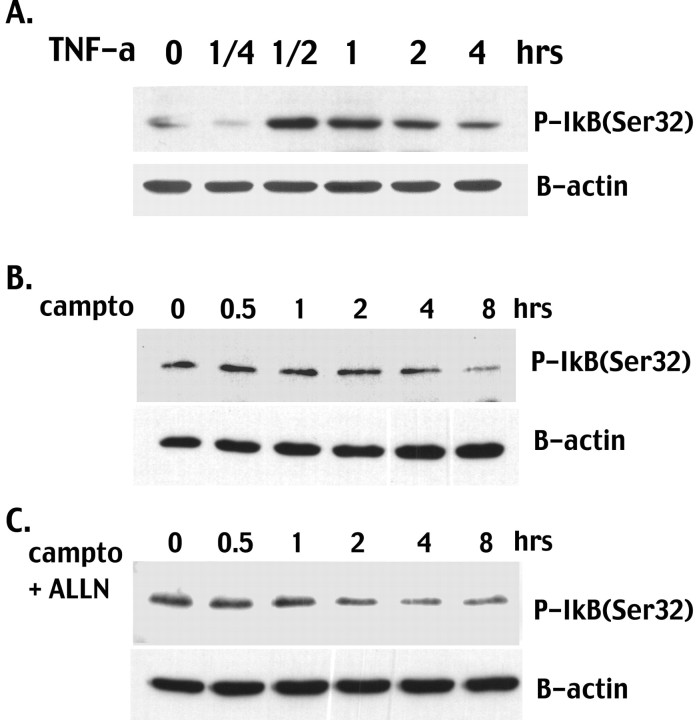
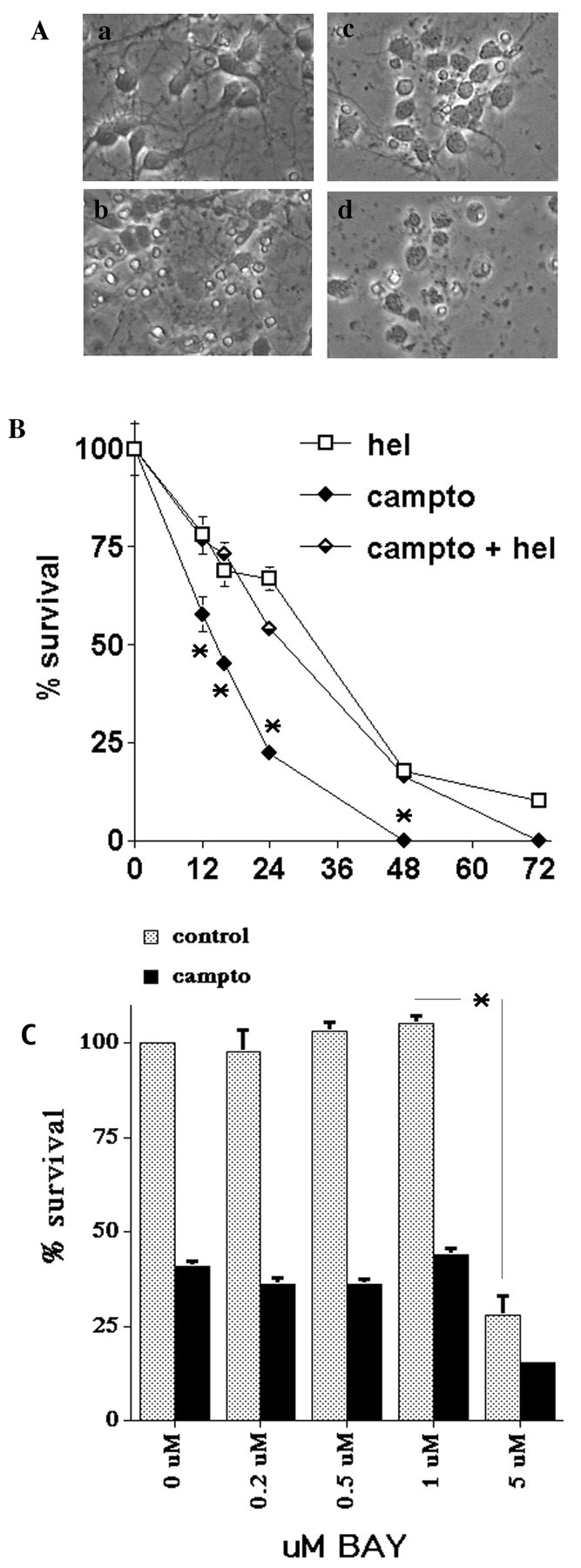
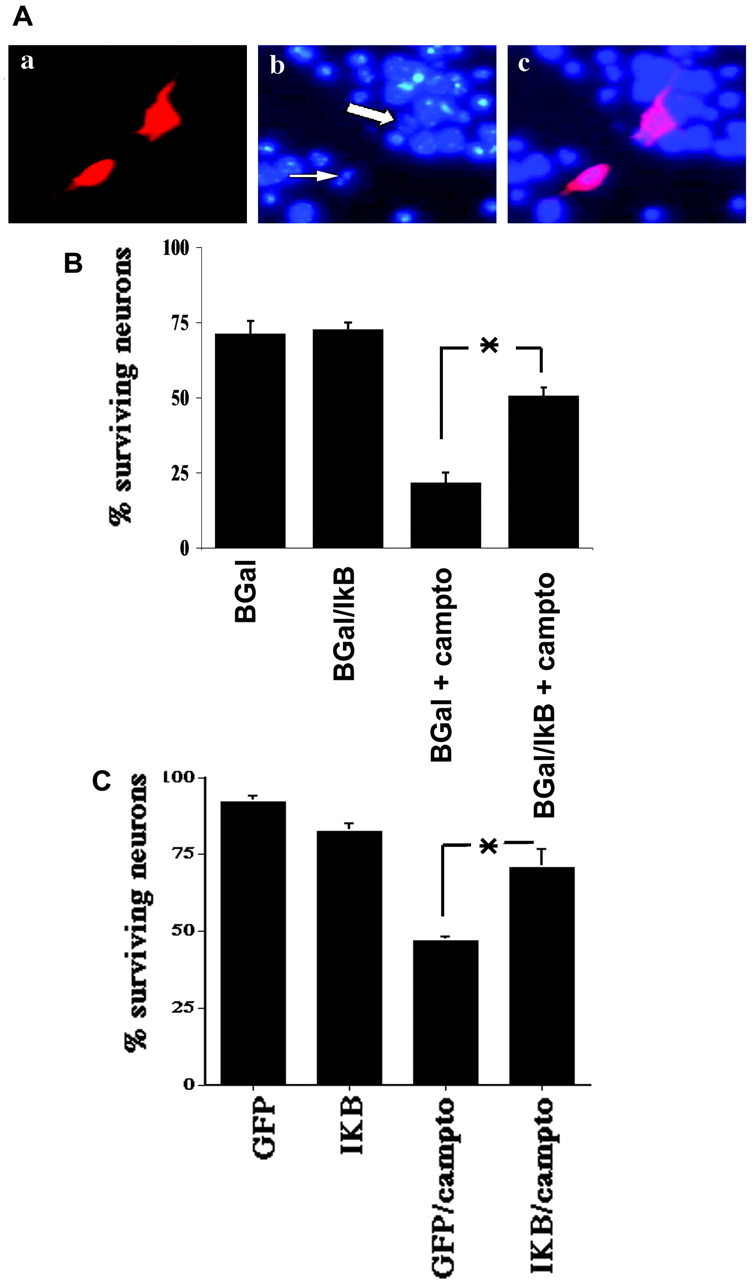
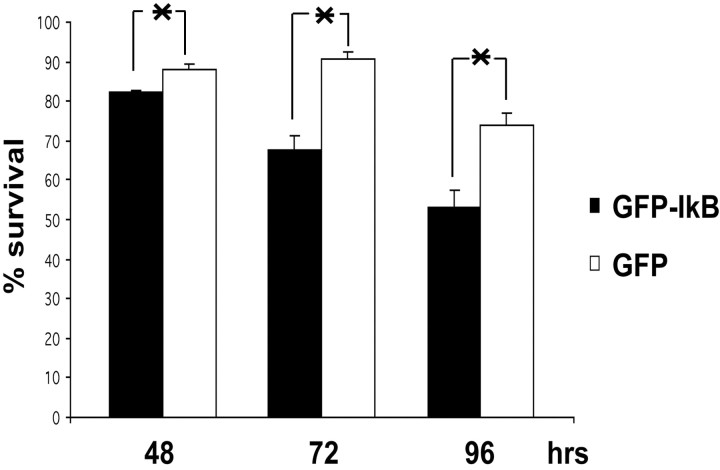
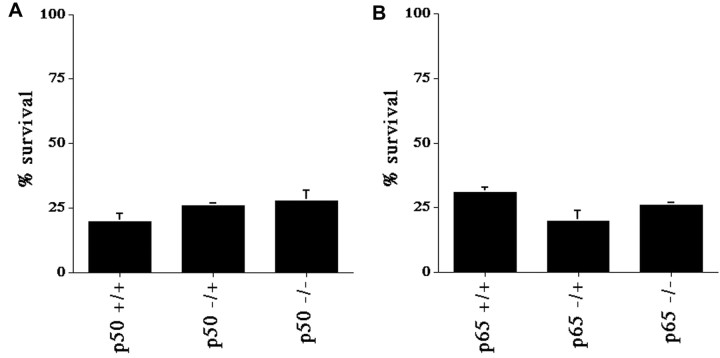
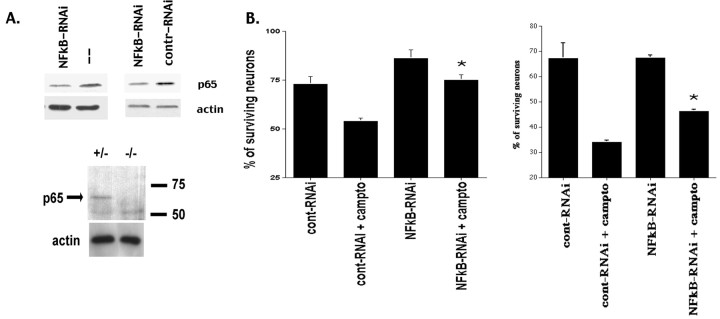
Previous studies have shown that DNA damage-evoked death of primary cortical neurons occurs in a p53 and cyclin-dependent kinase-dependent (CDK) manner. The manner by which these signals modulate death is unclear. Nuclear factor-kappaB (NF-kappaB) is a group of transcription factors that potentially interact with these pathways. Presently, we show that NF-kappaB is activated shortly after induction of DNA damage in a manner independent of the classic IkappaB kinase (IKK) activation pathway, CDKs, ATM, and p53. Acute inhibition of NF-kappaB via expression of a stable IkappaB mutant, downregulation of the p65 NF-kappaB subunit by RNA interference (RNAi), or pharmacological NF-kappaB inhibitors significantly protected against DNA damage-induced neuronal death. NF-kappaB inhibition also reduced p53 transcripts and p53 activity as measured by the p53-inducible messages, Puma and Noxa, implicating the p53 tumor suppressor in the mechanism of NF-kappaB-mediated neuronal death. Importantly, p53 expression still induces death in the presence of NF-kappaB inhibition, indicating that p53 acts downstream of NF-kappaB. Interestingly, neurons cultured from p65 or p50 NF-kappaB-deficient mice were not resistant to death and did not show diminished p53 activity, suggesting compensatory processes attributable to germline deficiencies, which allow p53 activation still to occur. In contrast to acute NF-kappaB inhibition, prolonged NF-kappaB inhibition caused neuronal death in the absence of DNA damage. These results uniquely define a signaling paradigm by which NF-kappaB serves both an acute p53-dependent pro-apoptotic function in the presence of DNA damage and an anti-apoptotic function in untreated normal neurons.
Figures
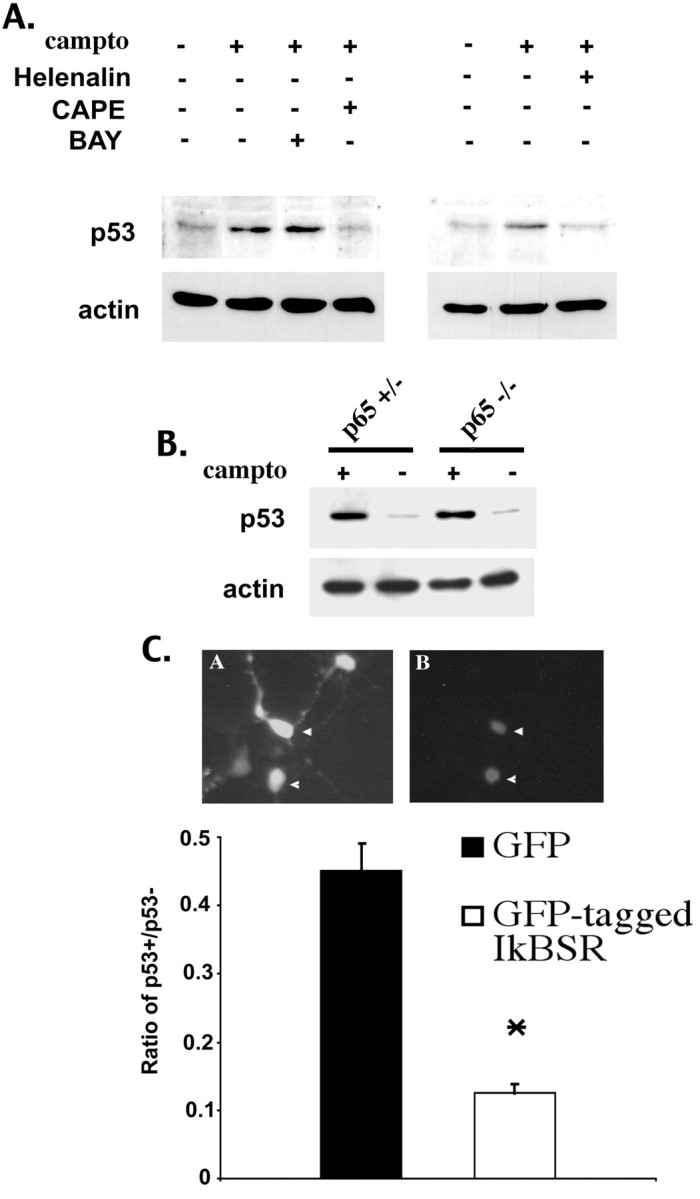
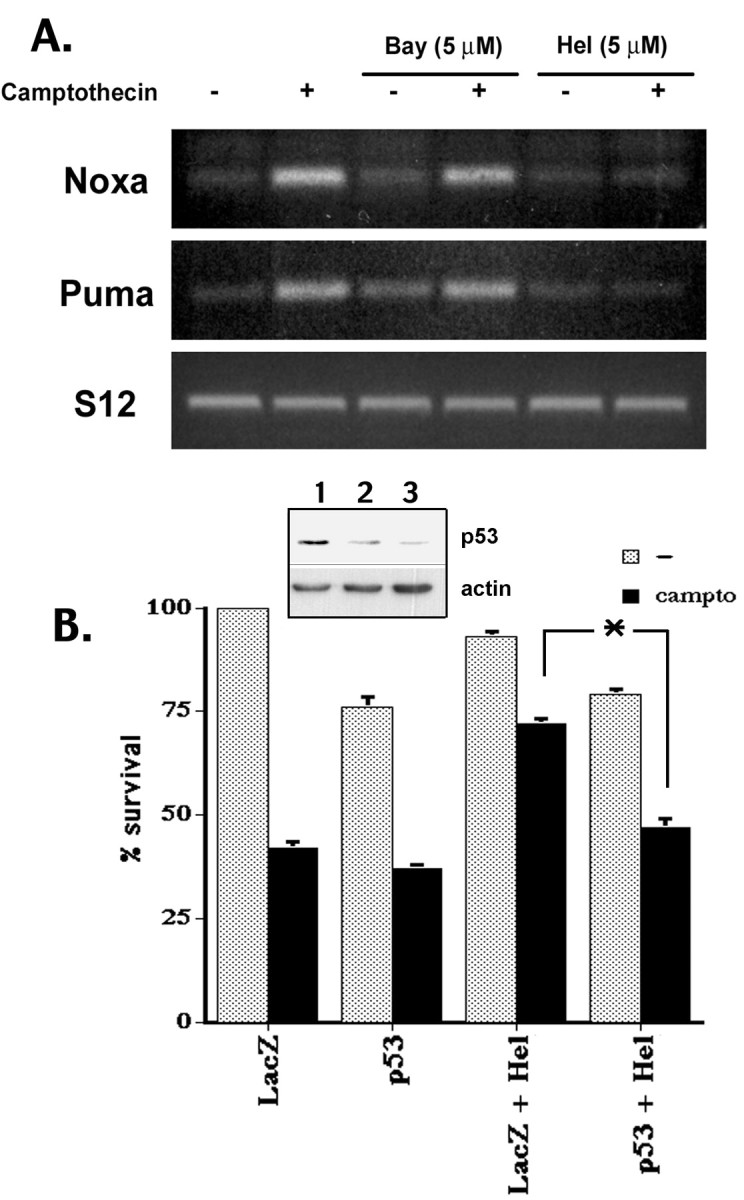
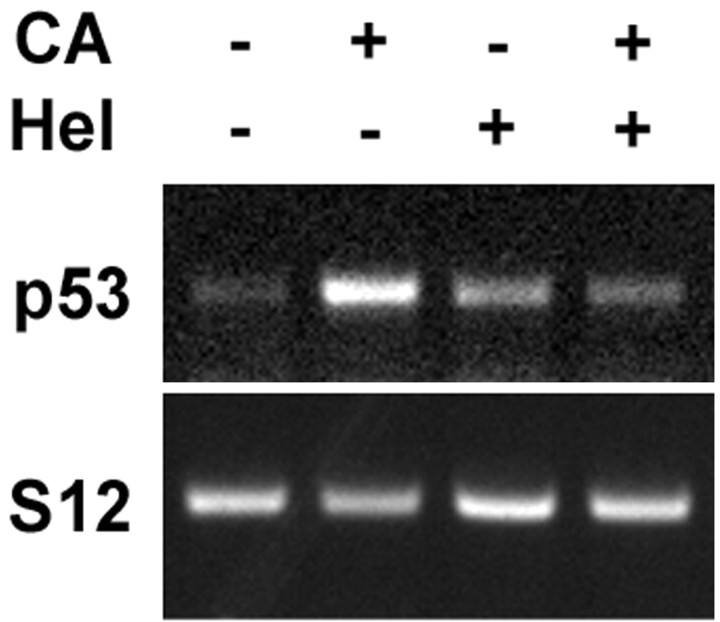
References
-
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B (1997) Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 69: 1196–1203. - PubMed
-
- Bales KR, Du Y, Dodel RC, Yan GM, Hamilton-Byrd E, Paul SM (1998) The NF-kappaB/Rel family of proteins mediates Aβ-induced neurotoxicity and glial activation. Brain Res Mol Brain Res 57: 63–72. - PubMed
-
- Barkett M, Gilmore TD (1999) Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18: 6910–6924. - PubMed
-
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T (1998) Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol 8: 1395–1398. - PubMed
-
- Benoit V, Hellin AC, Huygen S, Gielen J, Bours V, Merville MP (2000) Additive effect between NF-kappaB subunits and p53 protein for transcriptional activation of human p53 promoter. Oncogene 19: 4787–4794. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous