

NFI-Ski interactions mediate transforming growth factor beta modulation of human papillomavirus type 16 early gene expression
- PMID: 15047811
- PMCID: PMC374275
- DOI: 10.1128/jvi.78.8.3953-3964.2004
NFI-Ski interactions mediate transforming growth factor beta modulation of human papillomavirus type 16 early gene expression
Abstract
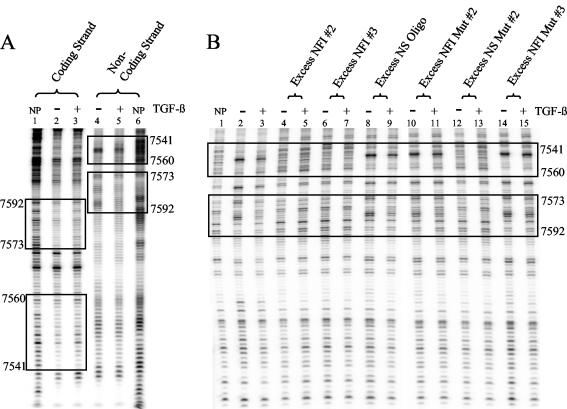
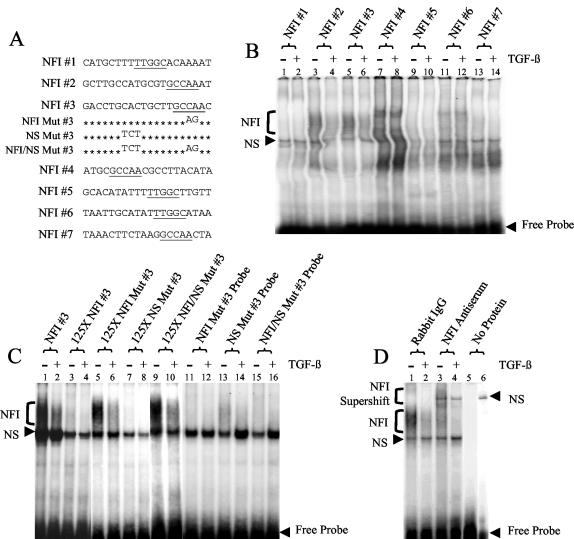
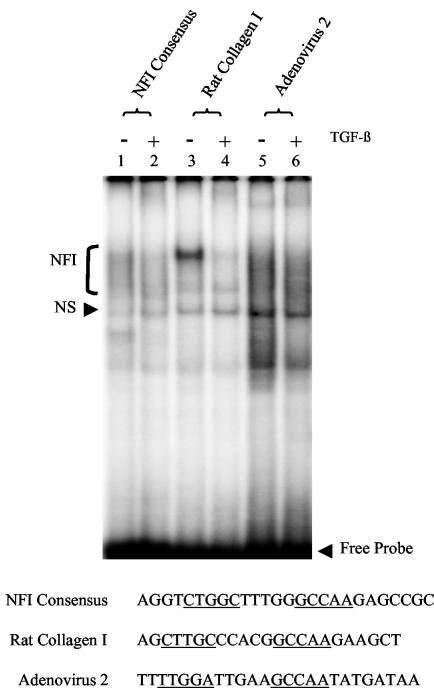
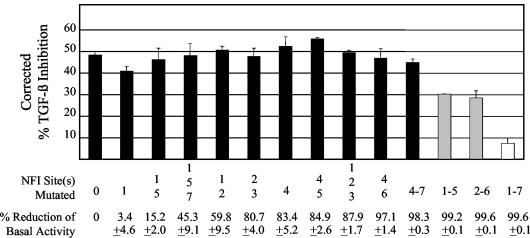
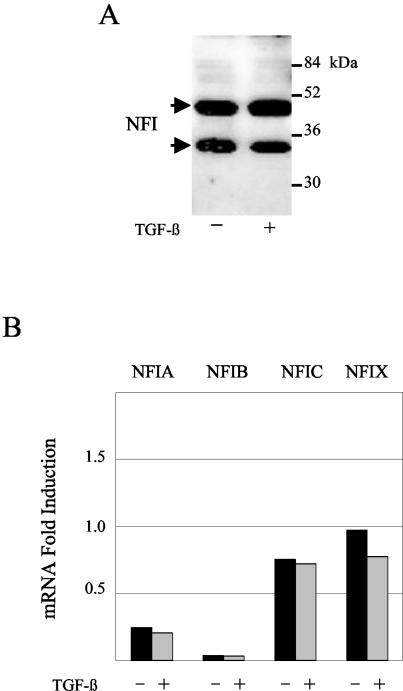
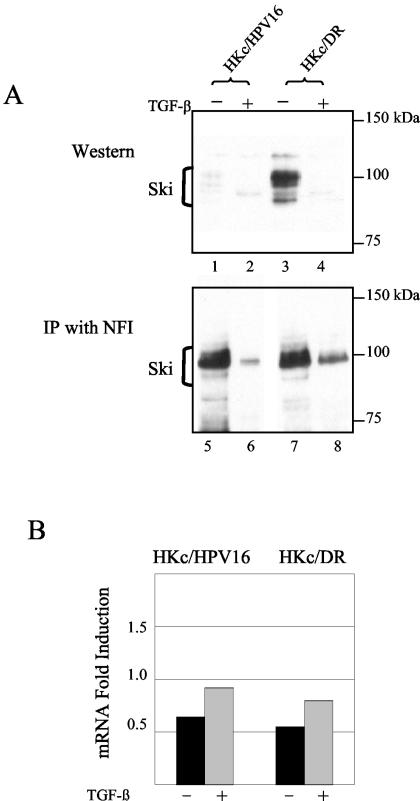
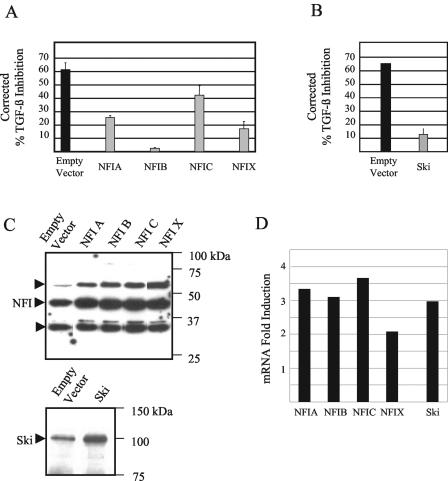
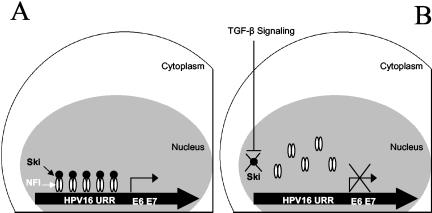
Human papillomaviruses (HPVs) are present in virtually all cervical cancers. An important step in the development of malignant disease, including cervical cancer, involves a loss of sensitivity to transforming growth factor beta (TGF-beta). HPV type 16 (HPV16) early gene expression, including that of the E6 and E7 oncoprotein genes, is under the control of the upstream regulatory region (URR), and E6 and E7 expression in HPV16-immortalized human epithelial cells is inhibited at the transcriptional level by TGF-beta. While the URR contains a myriad of transcription factor binding sites, including seven binding sites for nuclear factor I (NFI), the specific sequences within the URR or the transcription factors responsible for TGF-beta modulation of the URR remain unknown. To identify potential transcription factors and binding sites involved in TGF-beta modulation of the URR, we performed DNase I footprint analysis on the HPV16 URR using nuclear extracts from TGF-beta-sensitive HPV16-immortalized human keratinocytes (HKc/HPV16) treated with and without TGF-beta. Differentially protected regions were found to be located around NFI binding sites. Electrophoretic mobility shift assays, using the NFI binding sites as probes, showed decreased binding upon TGF-beta treatment. This decrease in binding was not due to reduced NFI protein or NFI mRNA levels. Mutational analysis of individual and multiple NFI binding sites in the URR defined their role in TGF-beta sensitivity of the promoter. Overexpression of the NFI family members in HKc/HPV16 decreased the ability of TGF-beta to inhibit the URR. Since the oncoprotein Ski has been shown to interact with and increase the transcriptional activity of NFI and since cellular Ski levels are decreased by TGF-beta treatment, we explored the possibility that Ski may provide a link between TGF-beta signaling and NFI activity. Anti-NFI antibodies coimmunoprecipitated endogenous Ski in nuclear extracts from HKc/HPV16, confirming that NFI and Ski interact in these cells. Ski levels dramatically decreased upon TGF-beta treatment of HKc/HPV16, and overexpression of Ski eliminated the ability of TGF-beta to inhibit the URR. Based on these studies, we propose that TGF-beta inhibition of HPV16 early gene expression is mediated by a decrease in Ski levels, which in turn dramatically reduces NFI activity.
Figures

References
-
- Berger, D. H., X. H. Feng, J. Yao, D. Saha, R. D. Beauchamp, and X. Lin. 2002. Resistance to transforming growth factor-beta occurs in the presence of normal Smad activation. Surgery 132:310-316. - PubMed
-
- Borger, D. R., Y. Mi, G. Geslani, L. L. Zyzak, A. Batova, T. S. Engin, L. Pirisi, and K. E. Creek. 2000. Retinoic acid resistance at late stages of human papillomavirus type 16-mediated transformation of human keratinocytes arises despite intact retinoid signaling and is due to a loss of sensitivity to transforming growth factor-beta. Virology 270:397-407. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
