

Structural and functional analysis of Mre11-3
- PMID: 15047855
- PMCID: PMC390353
- DOI: 10.1093/nar/gkh343
Structural and functional analysis of Mre11-3
Abstract
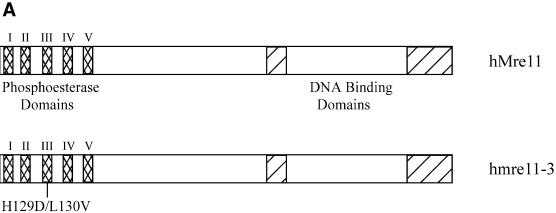
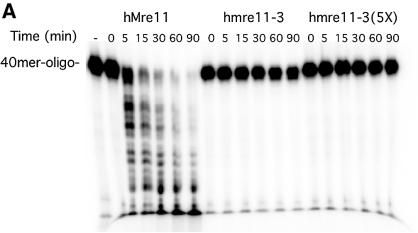
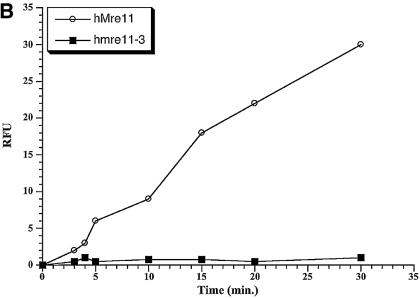
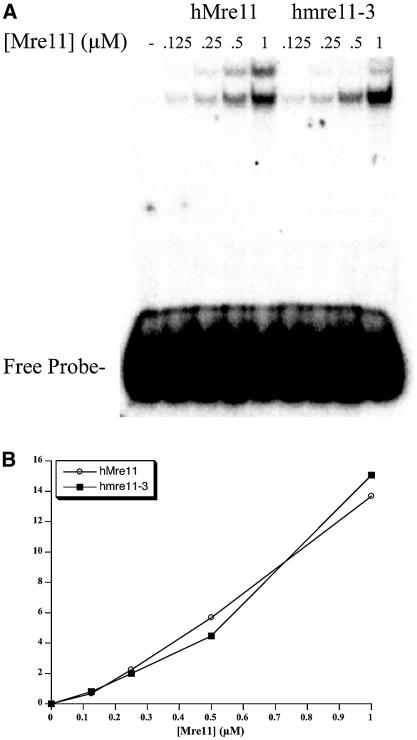
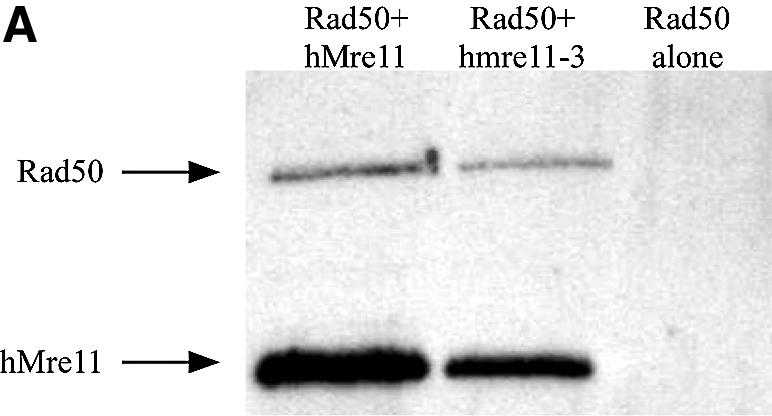
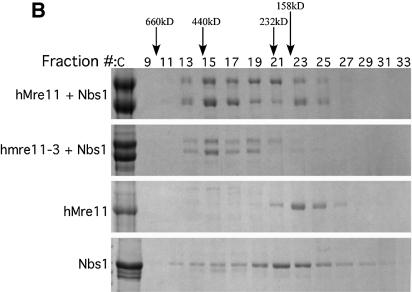
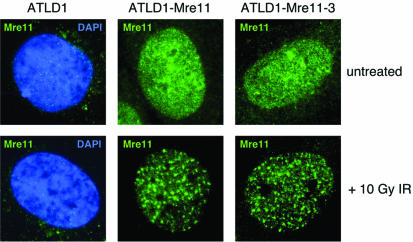
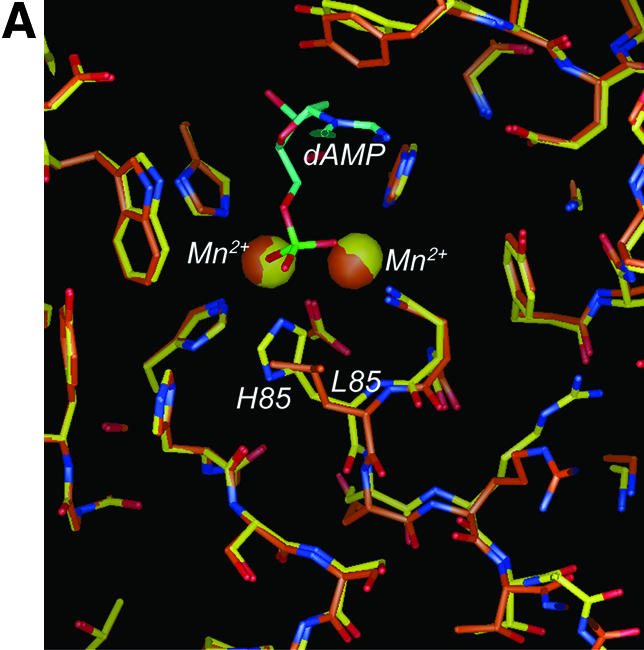
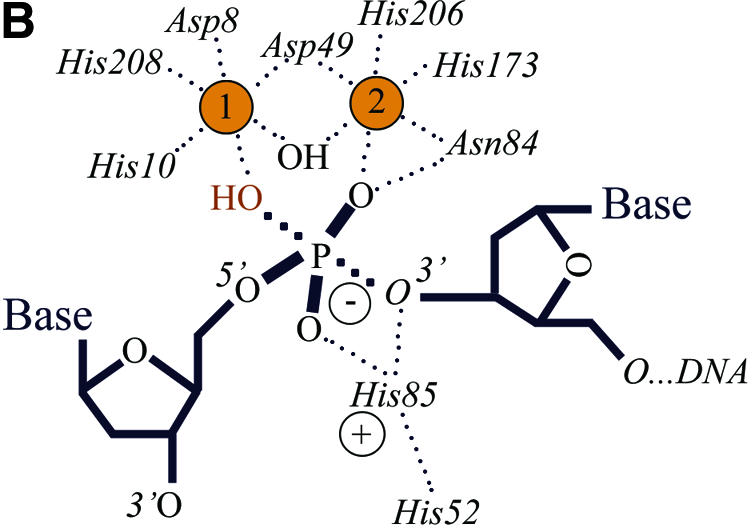
The Mre11, Rad50 and Nbs1 proteins make up the conserved multi-functional Mre11 (MRN) complex involved in multiple, critical DNA metabolic processes including double-strand break repair and telomere maintenance. The Mre11 protein is a nuclease with broad substrate recognition, but MRN-dependent processes requiring the nuclease activity are not clearly defined. Here, we report the functional and structural characterization of a nuclease-deficient Mre11 protein termed mre11-3. Importantly, the hmre11-3 protein has wild-type ability to bind DNA, Rad50 and Nbs1; however, nuclease activity was completely abrogated. When expressed in cell lines from patients with ataxia telangiectasia-like disorder (ATLD), hmre11-3 restored the formation of ionizing radiation-induced foci. Consistent with the biochemical results, the 2.3 A crystal structure of mre11-3 from Pyrococcus furiosus revealed an active site structure with a wild-type-like metal-binding environment. The structural analysis of the H85L mutation provides a detailed molecular basis for the ability of mre11-3 to bind but not hydrolyze DNA. Together, these results establish that the mre11-3 protein provides an excellent system for dissecting nuclease-dependent and independent functions of the Mre11 complex.
Figures
References
-
- Critchlow S.E. and Jackson,S.P. (1998) DNA end-joining: from yeast to man. Trends Biochem. Sci., 23, 394–398. - PubMed
-
- Varon R., Vissinga,C., Platzer,M., Cerosaletti,K.M., Chrzanowska,K.H., Saar,K., Beckmann,G., Seemanova,E., Cooper,P.R., Nowak,N.J. et al. (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell, 93, 467–476. - PubMed
-
- Carney J.P., Maser,R.S., Olivares,H., Davis,E.M., Le Beau,M., Yates,J.R., 3rd, Hays,L., Morgan,W.F. and Petrini,J.H. (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell, 93, 477–486. - PubMed
-
- Haber J.E. (1998) The many interfaces of Mre11. Cell, 95, 583–586. - PubMed
-
- Stewart G.S., Maser,R.S., Stankovic,T., Bressan,D.A., Kaplan,M.I., Jaspers,N.G., Raams,A., Byrd,P.J., Petrini,J.H. and Taylor,A.M. (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell, 99, 577–587. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
