

Quantifying the relationship between co-expression, co-regulation and gene function
- PMID: 15053845
- PMCID: PMC375525
- DOI: 10.1186/1471-2105-5-18
Quantifying the relationship between co-expression, co-regulation and gene function
Abstract
Background: It is thought that genes with similar patterns of mRNA expression and genes with similar functions are likely to be regulated via the same mechanisms. It has been difficult to quantitatively test these hypotheses on a large scale because there has been no general way of determining whether genes share a common regulatory mechanism. Here we use data from a recent genome wide binding analysis in combination with mRNA expression data and existing functional annotations to quantify the likelihood that genes with varying degrees of similarity in mRNA expression profile or function will be bound by a common transcription factor.
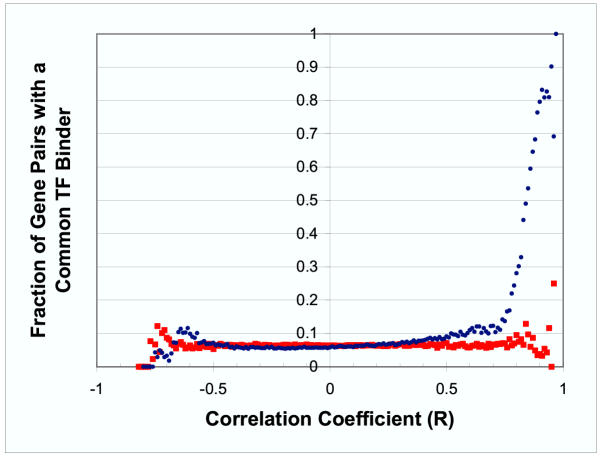
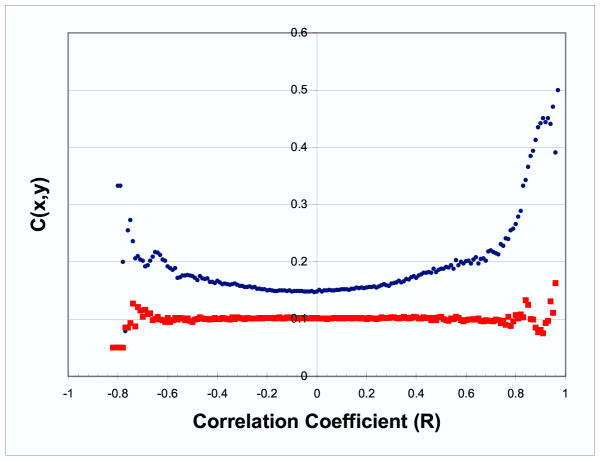
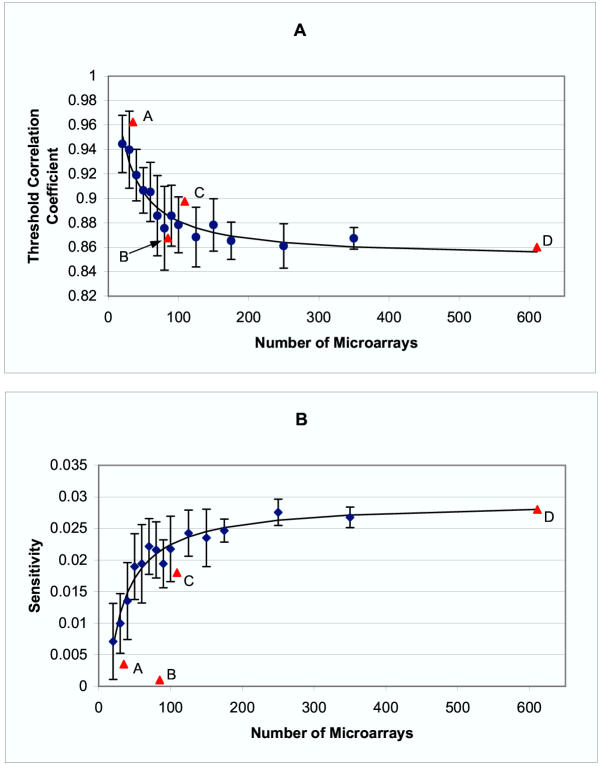
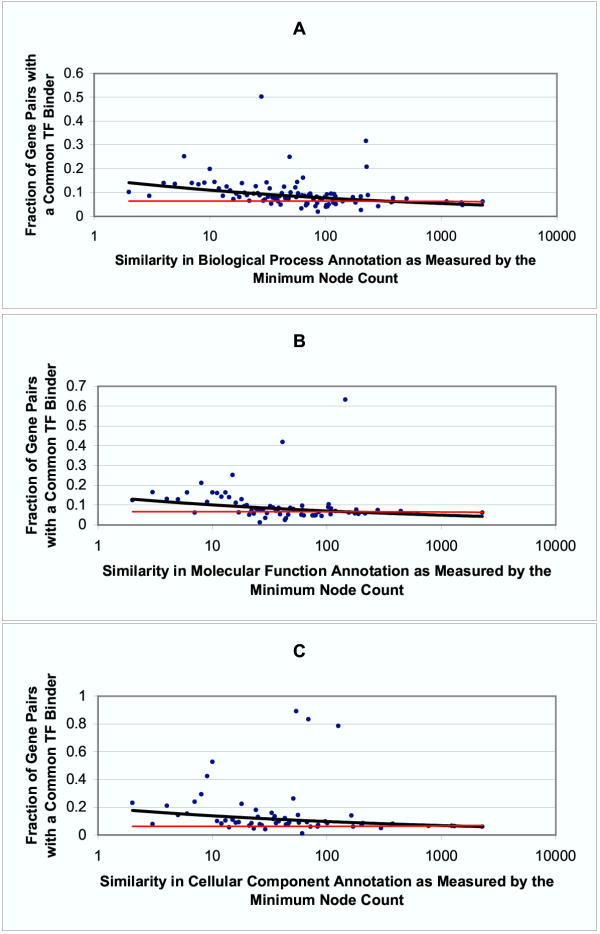
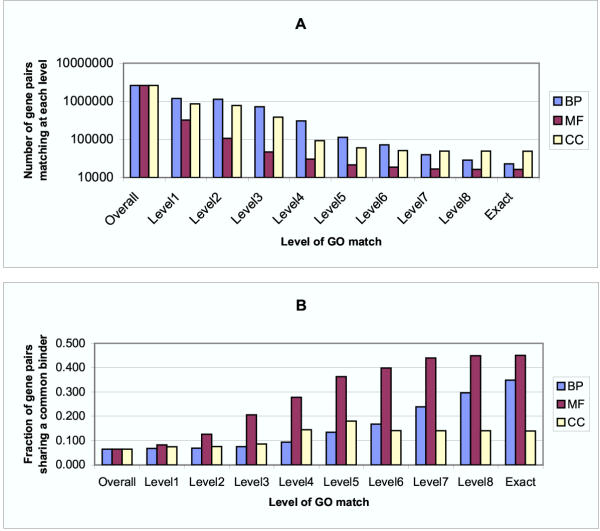
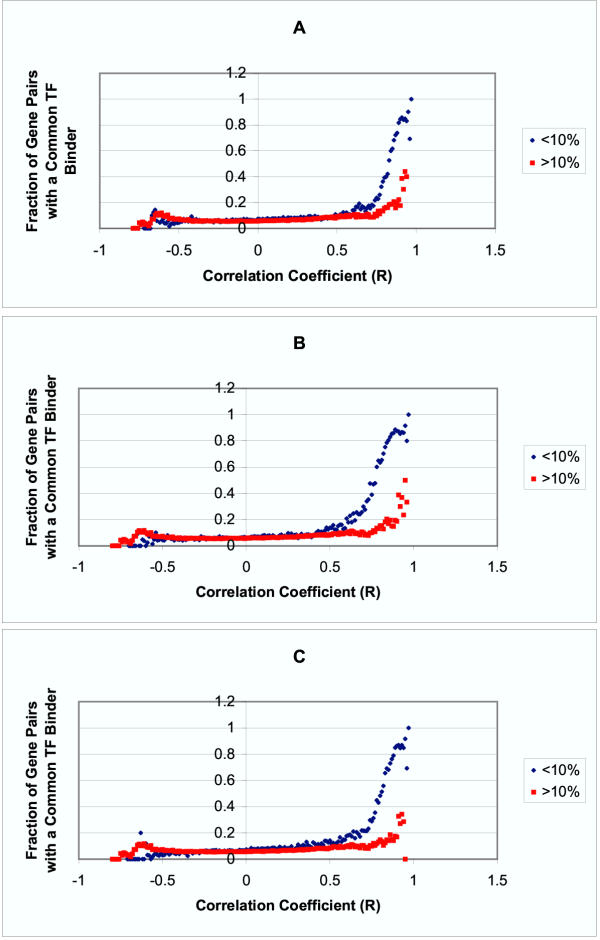
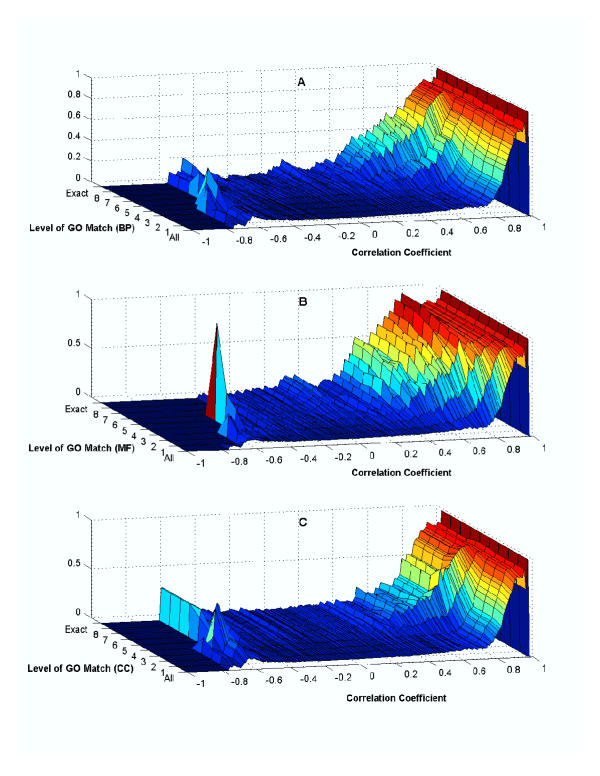
Results: Genes with strongly correlated mRNA expression profiles are more likely to have their promoter regions bound by a common transcription factor. This effect is present only at relatively high levels of expression similarity. In order for two genes to have a greater than 50% chance of sharing a common transcription factor binder, the correlation between their expression profiles (across the 611 microarrays used in our study) must be greater than 0.84. Genes with similar functional annotations are also more likely to be bound by a common transcription factor. Combining mRNA expression data with functional annotation results in a better predictive model than using either data source alone.
Conclusions: We demonstrate how mRNA expression data and functional annotations can be used together to estimate the probability that genes share a common regulatory mechanism. Existing microarray data and known functional annotations are sufficient to identify only a relatively small percentage of co-regulated genes.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
