

E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases
- PMID: 15057284
- PMCID: PMC394229
- DOI: 10.1038/sj.emboj.7600136
E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases
Abstract
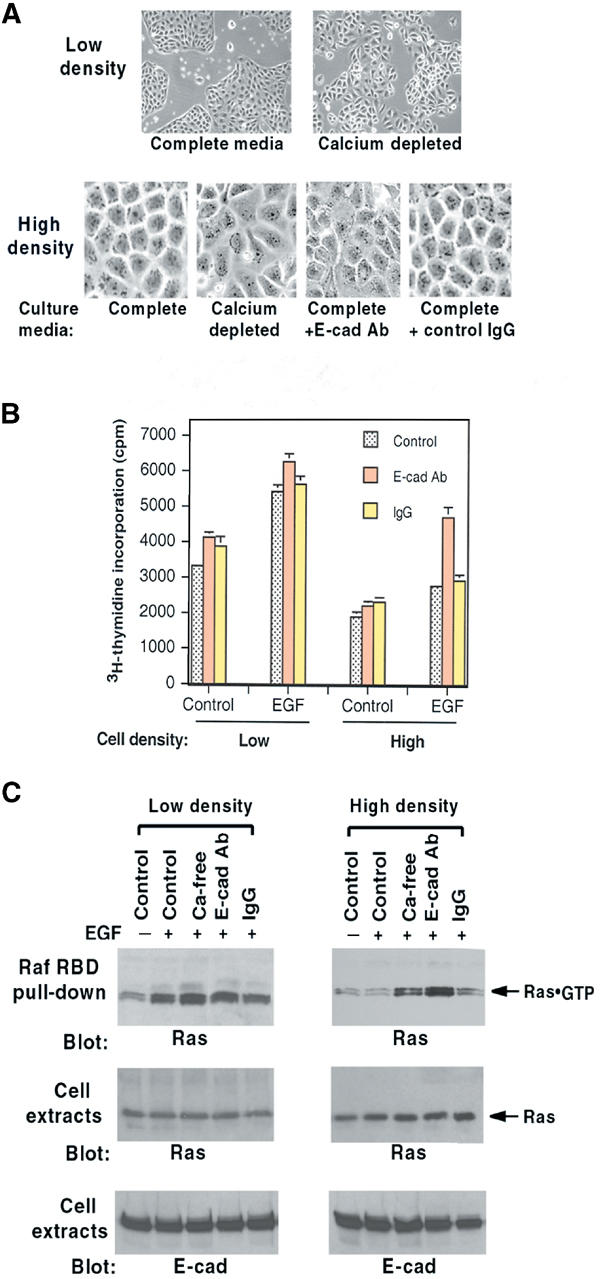
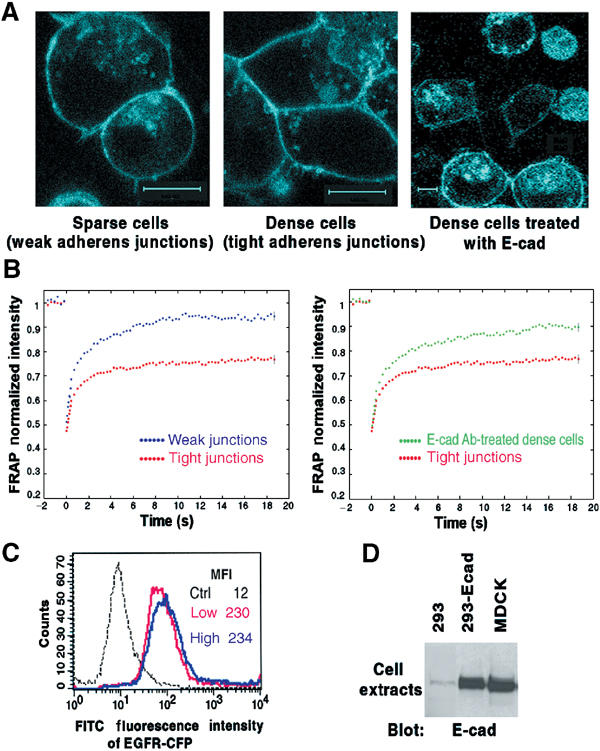
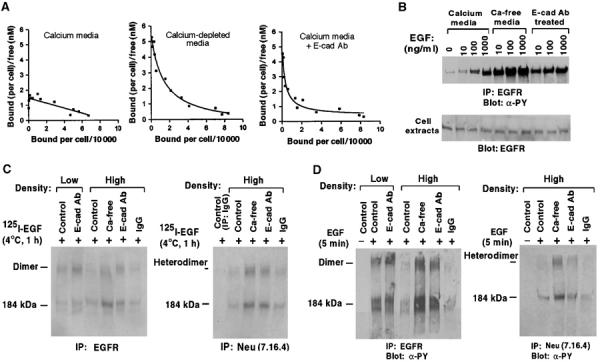
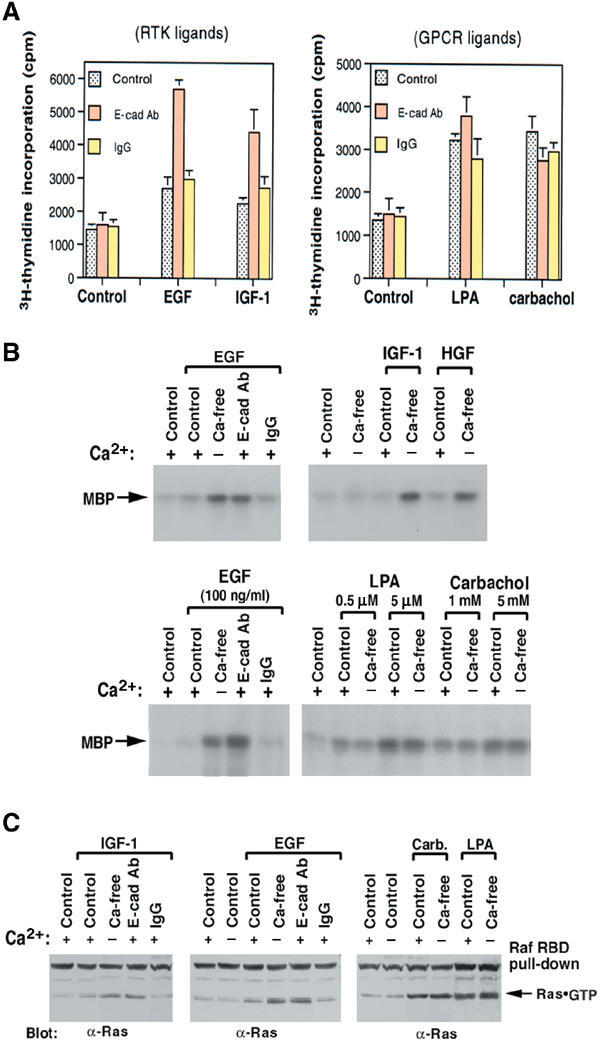
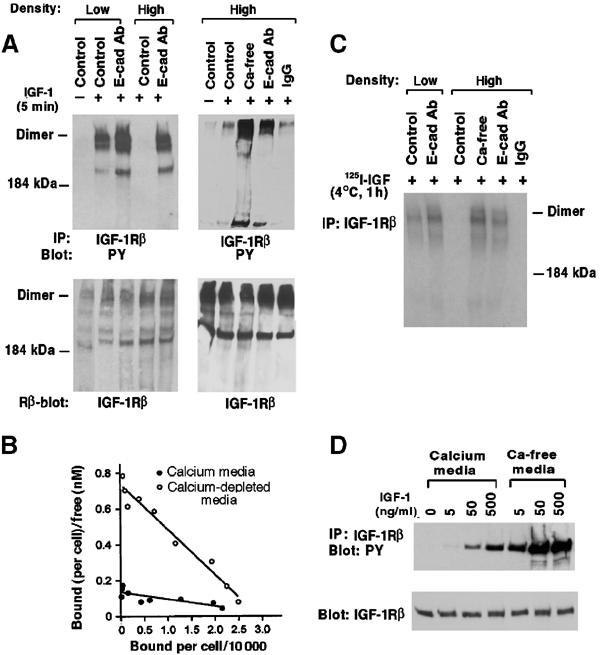
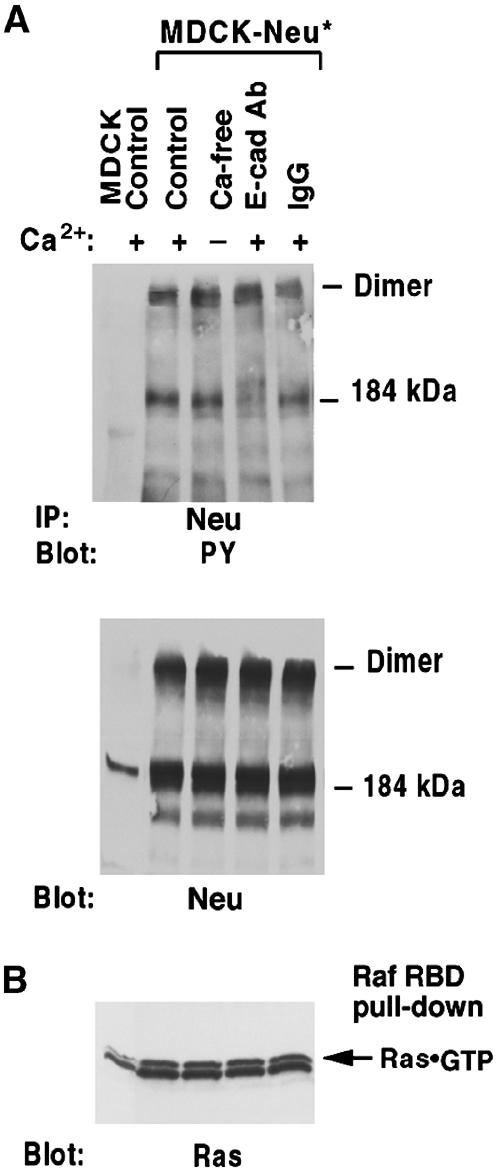
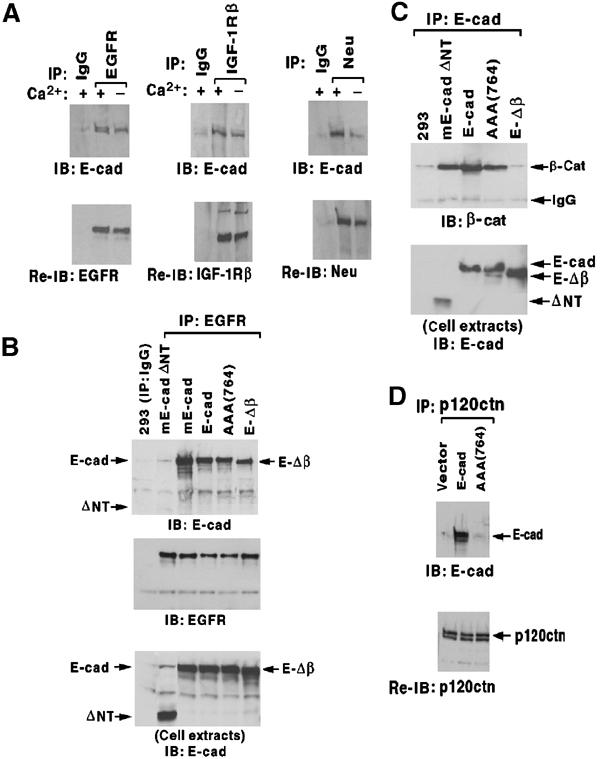
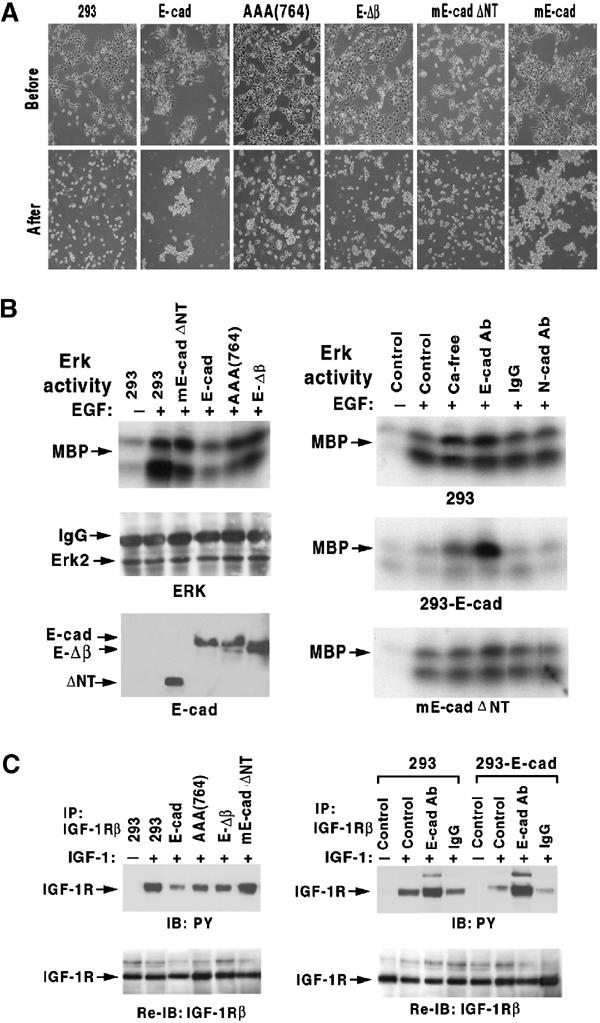
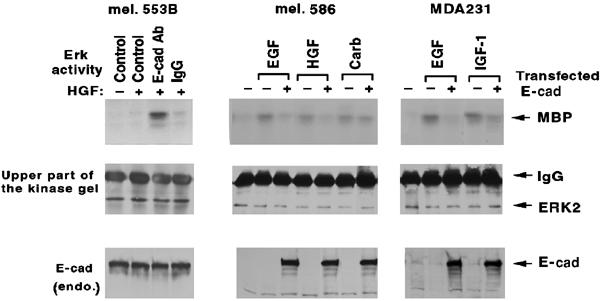
E-cadherin is an essential adhesion protein as well as a tumor suppressor that is silenced in many cancers. Its adhesion-dependent regulation of signaling has not been elucidated. We report that E-cadherin can negatively regulate, in an adhesion-dependent manner, the ligand-dependent activation of divergent classes of receptor tyrosine kinases (RTKs), by inhibiting their ligand-dependent activation in association with decreases in receptor mobility and in ligand-binding affinity. E-cadherin did not regulate a constitutively active mutant RTK (Neu*) or the ligand-dependent activation of LPA receptors or muscarinic receptors, which are two classes of G protein-coupled receptors. EGFR regulation by E-cadherin was associated with complex formation between EGFR and E-cadherin that depended on the extracellular domain of E-cadherin but was independent of beta-catenin binding or p120-catenin binding. Transfection of E-cadherin conferred negative RTK regulation to human melanoma and breast cancer lines with downregulated endogenous E-cadherin. Abrogation of E-cadherin regulation may contribute to the frequent ligand-dependent activation of RTK in tumors.
Figures
References
-
- Bargmann CI, Hung MC, Weinberg RA (1986) The neu oncogene encodes an epidermal growth factor receptor-related protein. Nature 319: 226–230 - PubMed
-
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355–365 - PubMed
-
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L (2002) C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296: 1308–1313 - PubMed
-
- Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98: 147–157 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
