

The regulatory content of intergenic DNA shapes genome architecture
- PMID: 15059258
- PMCID: PMC395784
- DOI: 10.1186/gb-2004-5-4-r25
The regulatory content of intergenic DNA shapes genome architecture
Abstract
Background: Factors affecting the organization and spacing of functionally unrelated genes in metazoan genomes are not well understood. Because of the vast size of a typical metazoan genome compared to known regulatory and protein-coding regions, functional DNA is generally considered to have a negligible impact on gene spacing and genome organization. In particular, it has been impossible to estimate the global impact, if any, of regulatory elements on genome architecture.
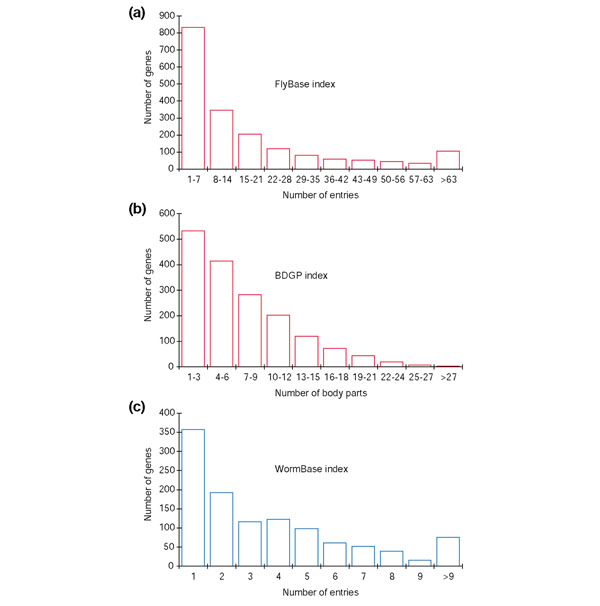
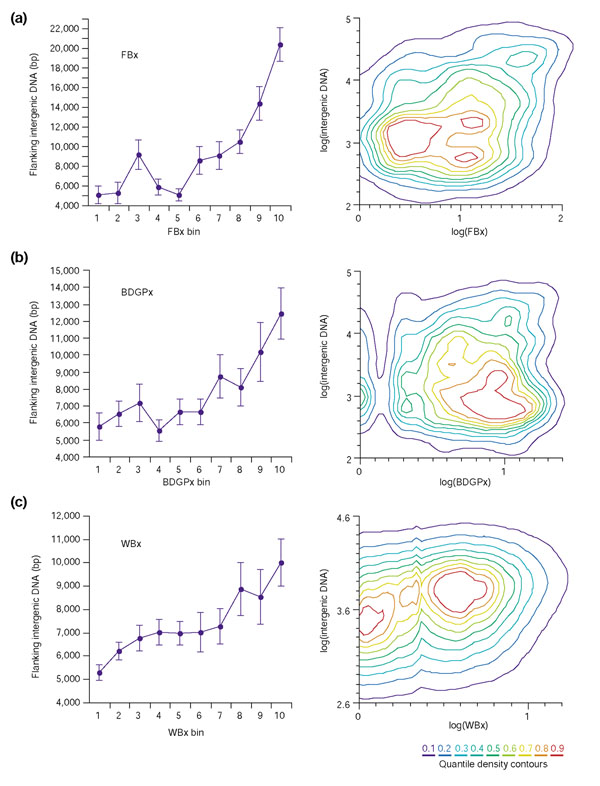
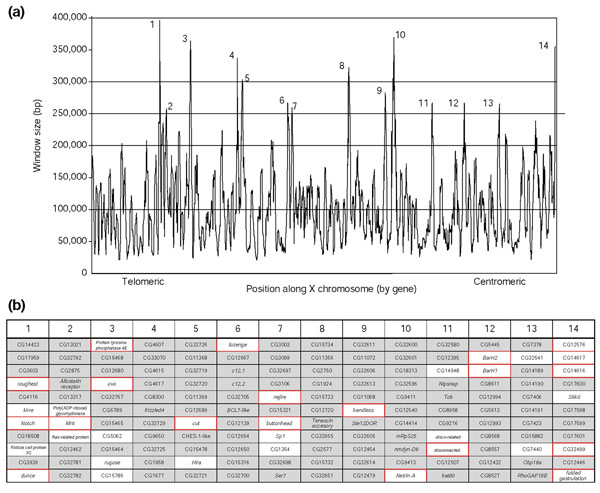
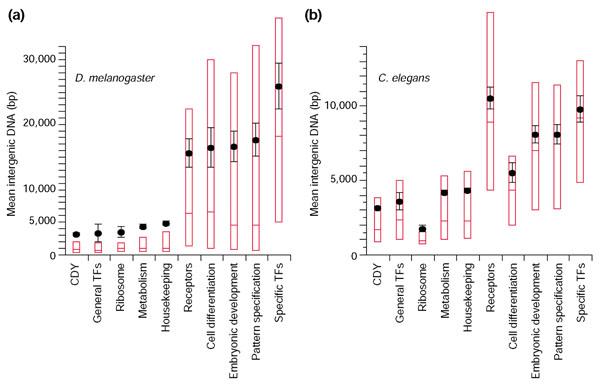
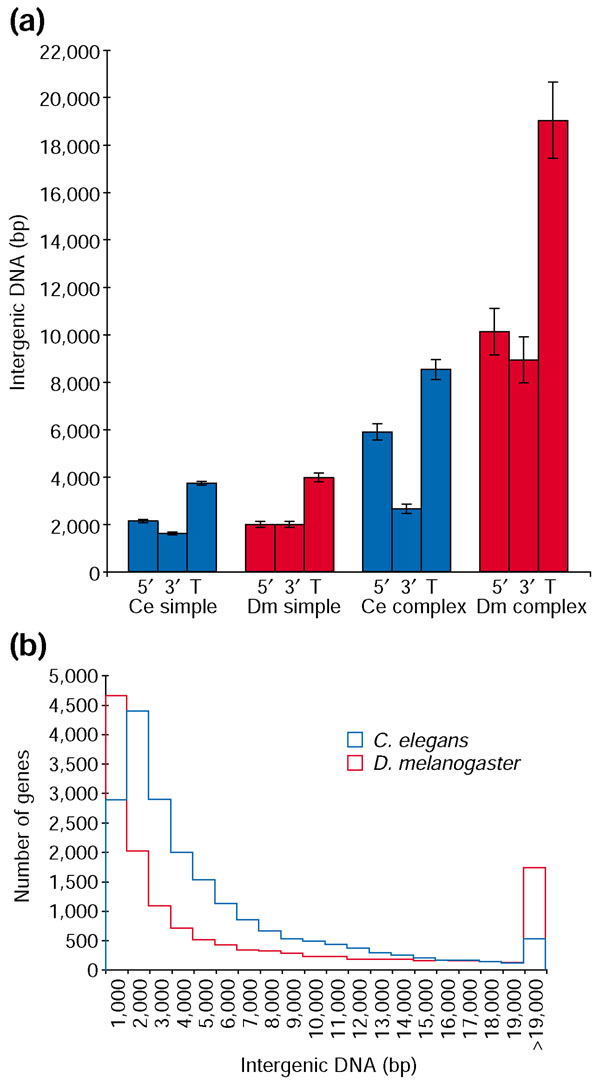
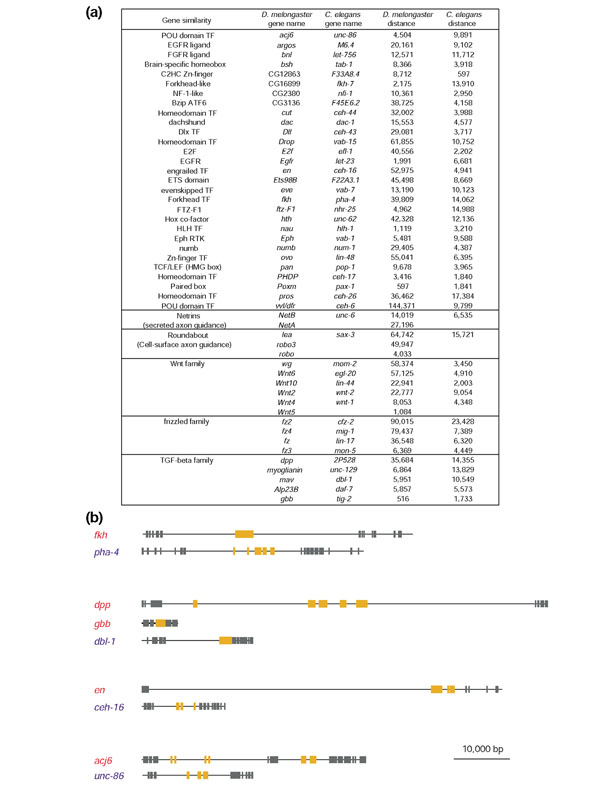
Results: To investigate this, we examined the relationship between regulatory complexity and gene spacing in Caenorhabditis elegans and Drosophila melanogaster. We found that gene density directly reflects local regulatory complexity, such that the amount of noncoding DNA between a gene and its nearest neighbors correlates positively with that gene's regulatory complexity. Genes with complex functions are flanked by significantly more noncoding DNA than genes with simple or housekeeping functions. Genes of low regulatory complexity are associated with approximately the same amount of noncoding DNA in D. melanogaster and C. elegans, while loci of high regulatory complexity are significantly larger in the more complex animal. Complex genes in C. elegans have larger 5' than 3' noncoding intervals, whereas those in D. melanogaster have roughly equivalent 5' and 3' noncoding intervals.
Conclusions: Intergenic distance, and hence genome architecture, is highly nonrandom. Rather, it is shaped by regulatory information contained in noncoding DNA. Our findings suggest that in compact genomes, the species-specific loss of nonfunctional DNA reveals a landscape of regulatory information by leaving a profile of functional DNA in its wake.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
