

MAVID: constrained ancestral alignment of multiple sequences
- PMID: 15060012
- PMCID: PMC383315
- DOI: 10.1101/gr.1960404
MAVID: constrained ancestral alignment of multiple sequences
Abstract
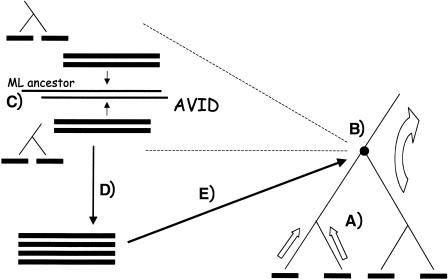
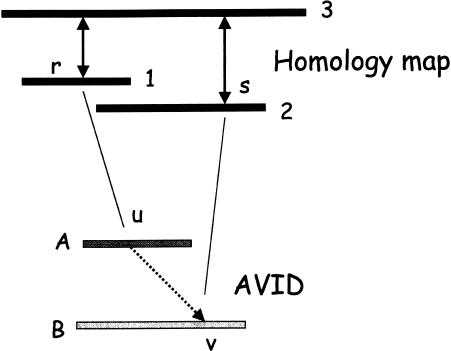
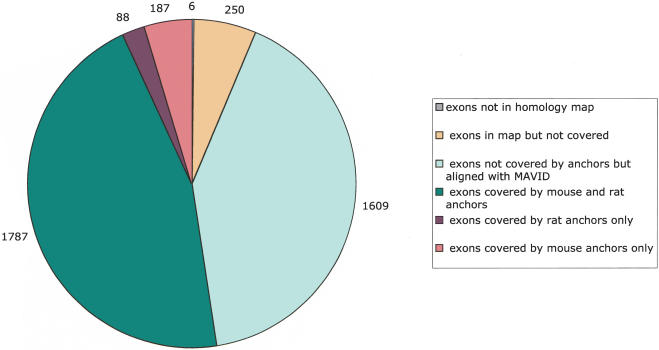
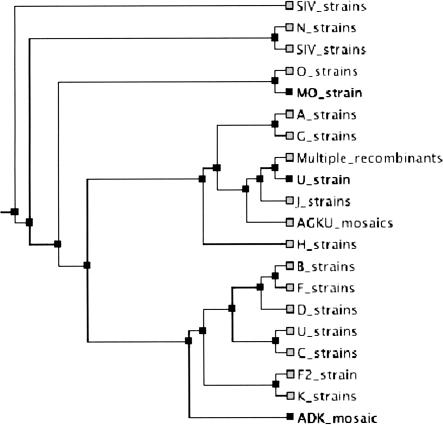
We describe a new global multiple-alignment program capable of aligning a large number of genomic regions. Our progressive-alignment approach incorporates the following ideas: maximum-likelihood inference of ancestral sequences, automatic guide-tree construction, protein-based anchoring of ab-initio gene predictions, and constraints derived from a global homology map of the sequences. We have implemented these ideas in the MAVID program, which is able to accurately align multiple genomic regions up to megabases long. MAVID is able to effectively align divergent sequences, as well as incomplete unfinished sequences. We demonstrate the capabilities of the program on the benchmark CFTR region, which consists of 1.8 Mb of human sequence and 20 orthologous regions in marsupials, birds, fish, and mammals. Finally, we describe two large MAVID alignments, an alignment of all the available HIV genomes and a multiple alignment of the entire human, mouse, and rat genomes.
Figures
References
-
- Boffelli, B., McAuliffe, J., Ovcharenko, D., Lewis, K.D., Ovcharenko, I., Pachter, L., and Rubin, E.M. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391-1394. - PubMed
-
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. - PubMed
WEB SITE REFERENCES
-
- http://www.nisc.nih.gov/; NIH Intramural Sequencing Center.
-
- http://hiv-web.lanl.gov/; LANL HIV Databases.
-
- http://baboon.math.berkeley.edu/mavid/; The MAVID Web server.
-
- http://baboon.math.berkeley.edu/mavid/data/; Supplemental Data.
-
- http://hanuman.math.berkeley.edu/kbrowser/; K-BROWSER.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous