

Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen
- PMID: 15066790
- PMCID: PMC383139
- DOI: 10.1128/AEM.70.4.1999-2012.2004
Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen
Erratum in
- Appl Environ Microbiol. 2008 Mar;74(6):1961
Abstract
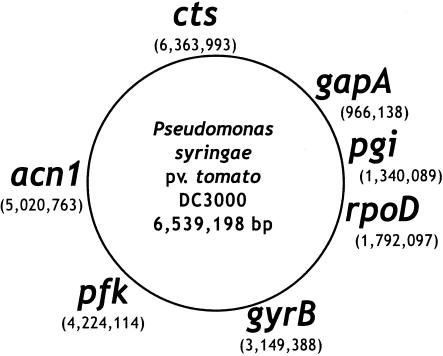
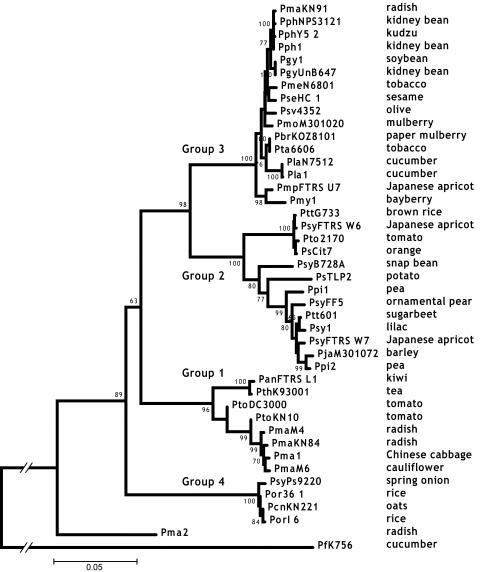
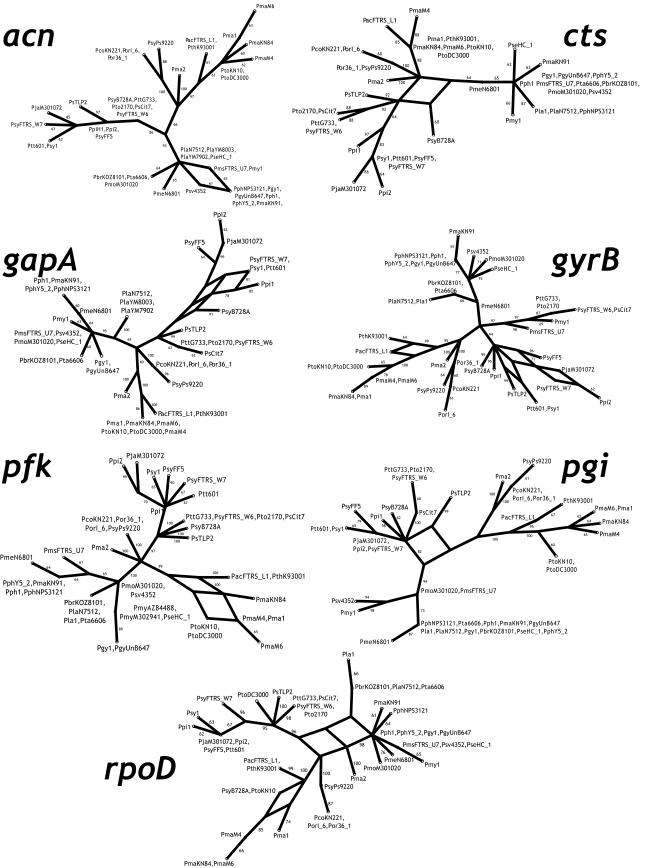
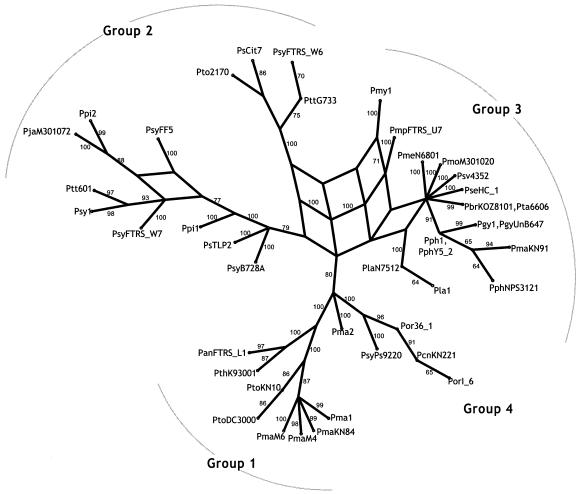
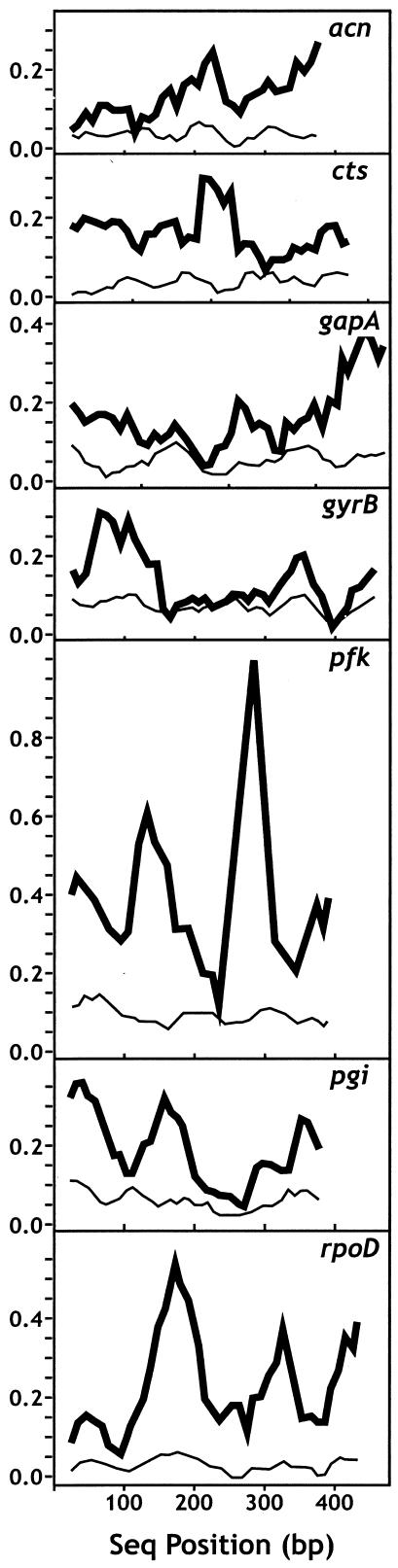
Pseudomonas syringae is a common foliar bacterium responsible for many important plant diseases. We studied the population structure and dynamics of the core genome of P. syringae via multilocus sequencing typing (MLST) of 60 strains, representing 21 pathovars and 2 nonpathogens, isolated from a variety of plant hosts. Seven housekeeping genes, dispersed around the P. syringae genome, were sequenced to obtain 400 to 500 nucleotides per gene. Forty unique sequence types were identified, with most strains falling into one of four major clades. Phylogenetic and maximum-likelihood analyses revealed a remarkable degree of congruence among the seven genes, indicating a common evolutionary history for the seven loci. MLST and population genetic analyses also found a very low level of recombination. Overall, mutation was found to be approximately four times more likely than recombination to change any single nucleotide. A skyline plot was used to study the demographic history of P. syringae. The species was found to have maintained a constant population size over time. Strains were also found to remain genetically homogeneous over many years, and when isolated from sites as widespread as the United States and Japan. An analysis of molecular variance found that host association explains only a small proportion of the total genetic variation in the sample. These analyses reveal that with respect to the core genome, P. syringae is a highly clonal and stable species that is endemic within plant populations, yet the genetic variation seen in these genes only weakly predicts host association.
Figures
References
-
- Awadalla, P. 2003. The evolutionary genomics of pathogen recombination. Nat. Rev. Genet. 4:50-60. - PubMed
-
- Bandelt, H. J., and A. W. Dress. 1992. Split decomposition: a new and useful approach to phylogenetic analysis of distance data. Mol. Phylogenet. Evol. 1:242-252. - PubMed
-
- Barker, F. K., and F. M. Lutzoni. 2002. The utility of the incongruence length difference test. Syst. Biol. 51:625-637. - PubMed
-
- Cunningham, C. W. 1997. Can three incongruence tests predict when data should be combined? Mol. Biol. Evol. 14:733-740. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
