

Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain
- PMID: 15069188
- PMCID: PMC395914
- DOI: 10.1073/pnas.0307565101
Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain
Abstract
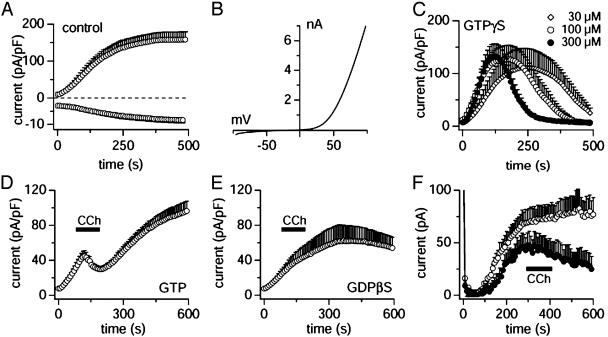
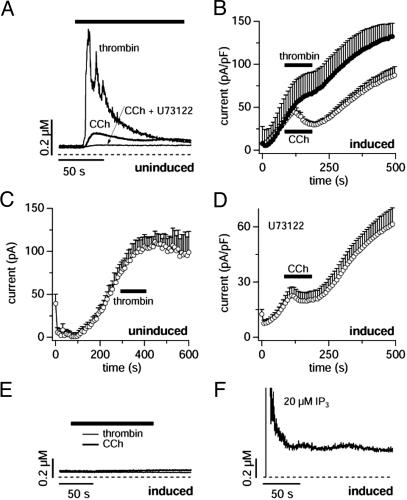
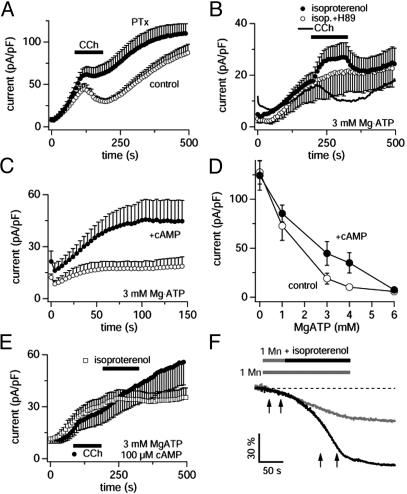
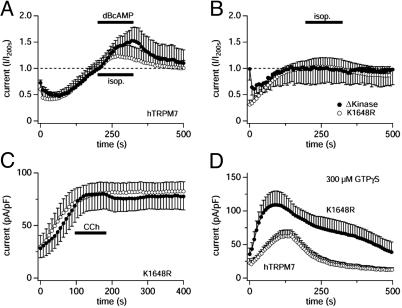
TRPM7 is a ubiquitously expressed and constitutively active divalent cation-selective ion channel, whose basal activity is regulated by intracellular levels of Mg(2+) and Mg.ATP. We have investigated receptor-mediated mechanisms that may actively regulate TRPM7 activity. We here report that TRPM7 currents are suppressed by intracellular GTPgammaS, suggesting the involvement of heterotrimeric G proteins. TRPM7 currents are also inhibited by stimulating endogenous muscarinic receptors, which is mediated by G(i) because the inhibitory effect is blunted by pertussis toxin. Conversely, stimulation of endogenous G(s)-coupled beta-adrenergic receptors potentiates TRPM7 currents, whereas G(q)-coupled thrombin receptors have little effect. Consistent with the involvement of G(s)/G(i) in controlling adenylyl cyclase activity, elevations of intracellular cAMP levels enhance TRPM7 activity and prevent receptor-mediated modulation of TRPM7 activity by muscarinic and adrenergic agonists. This cAMP-dependent effect requires the functional integrity of both protein kinase A (PKA) and the endogenous kinase domain of TRPM7 because cAMP-mediated effects are abolished when treating cells with the PKA inhibitors H89 or KT5720 as well as in cells expressing phosphotransferase-deficient TRPM7 constructs. These mutant channels are also much less susceptible to GTPgammaS-mediated inhibition, suggesting that the main regulatory effect occurs through G(i)- and G(s)-mediated changes in cAMP. Taken together, our results demonstrate that TRPM7 activity is up- and down-regulated through its endogenous kinase in a cAMP- and PKA-dependent manner.
Figures
References
-
- Clapham, D. E., Runnels, L. W. & Strubing, C. (2001) Nat. Rev. Neurosci. 2, 387–396. - PubMed
-
- Harteneck, C., Plant, T. D. & Schultz, G. (2000) Trends Neurosci. 23, 159–166. - PubMed
-
- Montell, C., Birnbaumer, L., Flockerzi, V., Bindels, R. J., Bruford, E. A., Caterina, M. J., Clapham, D. E., Harteneck, C., Heller, S., Julius, D., et al. (2002) Mol. Cell 9, 229–231. - PubMed
-
- Montell, C., Birnbaumer, L. & Flockerzi, V. (2002) Cell 108, 595–598. - PubMed
-
- Nadler, M. J., Hermosura, M. C., Inabe, K., Perraud, A. L., Zhu, Q., Stokes, A. J., Kurosaki, T., Kinet, J. P., Penner, R., Scharenberg, A. M., et al. (2001) Nature 411, 590–595. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
