

A Hidden Markov Model method, capable of predicting and discriminating beta-barrel outer membrane proteins
- PMID: 15070403
- PMCID: PMC385222
- DOI: 10.1186/1471-2105-5-29
A Hidden Markov Model method, capable of predicting and discriminating beta-barrel outer membrane proteins
Abstract
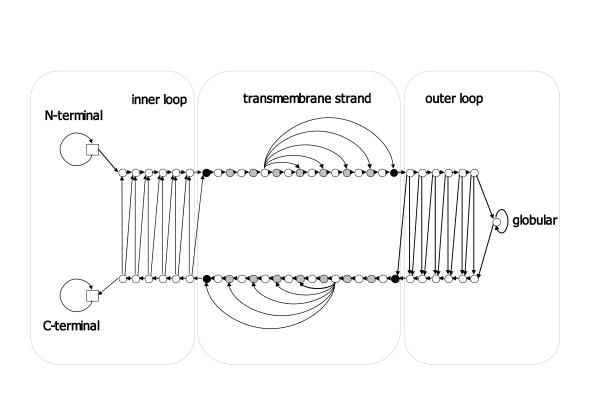
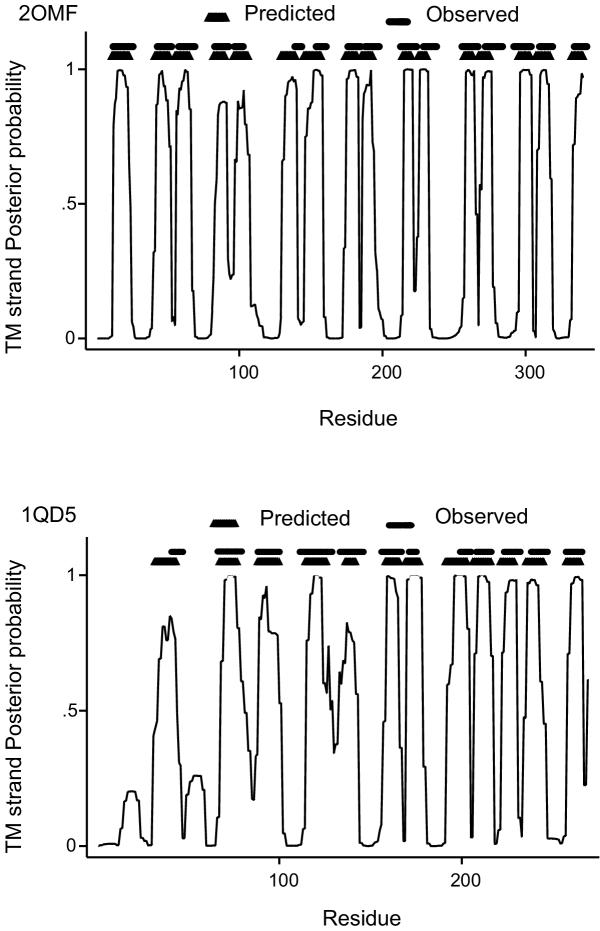
Background: Integral membrane proteins constitute about 20-30% of all proteins in the fully sequenced genomes. They come in two structural classes, the alpha-helical and the beta-barrel membrane proteins, demonstrating different physicochemical characteristics, structure and localization. While transmembrane segment prediction for the alpha-helical integral membrane proteins appears to be an easy task nowadays, the same is much more difficult for the beta-barrel membrane proteins. We developed a method, based on a Hidden Markov Model, capable of predicting the transmembrane beta-strands of the outer membrane proteins of gram-negative bacteria, and discriminating those from water-soluble proteins in large datasets. The model is trained in a discriminative manner, aiming at maximizing the probability of correct predictions rather than the likelihood of the sequences.
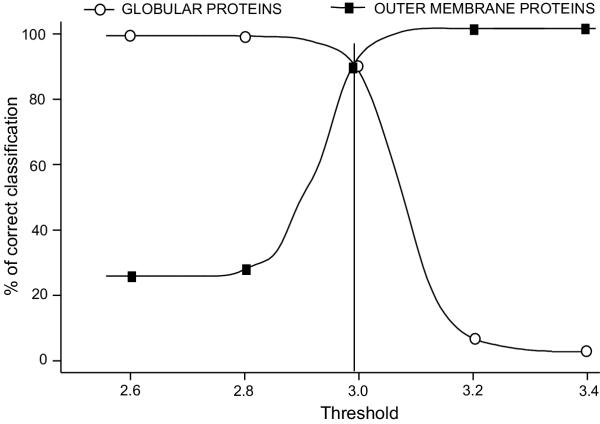
Results: The training has been performed on a non-redundant database of 14 outer membrane proteins with structures known at atomic resolution; it has been tested with a jacknife procedure, yielding a per residue accuracy of 84.2% and a correlation coefficient of 0.72, whereas for the self-consistency test the per residue accuracy was 88.1% and the correlation coefficient 0.824. The total number of correctly predicted topologies is 10 out of 14 in the self-consistency test, and 9 out of 14 in the jacknife. Furthermore, the model is capable of discriminating outer membrane from water-soluble proteins in large-scale applications, with a success rate of 88.8% and 89.2% for the correct classification of outer membrane and water-soluble proteins respectively, the highest rates obtained in the literature. That test has been performed independently on a set of known outer membrane proteins with low sequence identity with each other and also with the proteins of the training set.
Conclusion: Based on the above, we developed a strategy, that enabled us to screen the entire proteome of E. coli for outer membrane proteins. The results were satisfactory, thus the method presented here appears to be suitable for screening entire proteomes for the discovery of novel outer membrane proteins. A web interface available for non-commercial users is located at: http://bioinformatics.biol.uoa.gr/PRED-TMBB, and it is the only freely available HMM-based predictor for beta-barrel outer membrane protein topology.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
