

Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries
- PMID: 15070753
- PMCID: PMC384782
- DOI: 10.1073/pnas.0400678101
Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries
Abstract
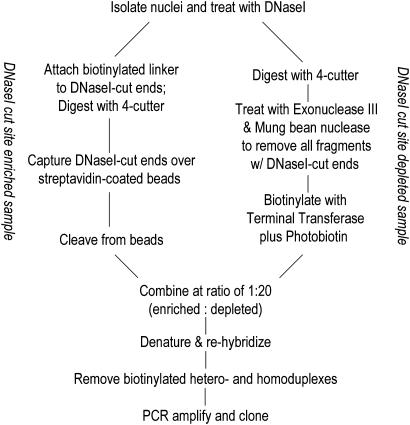
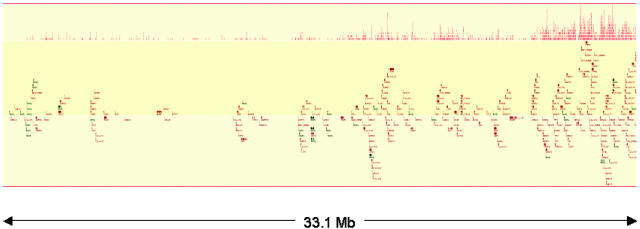
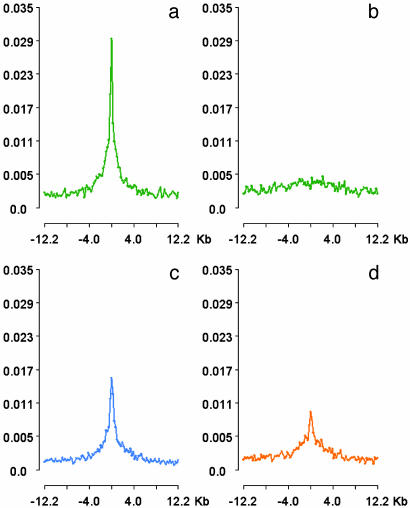
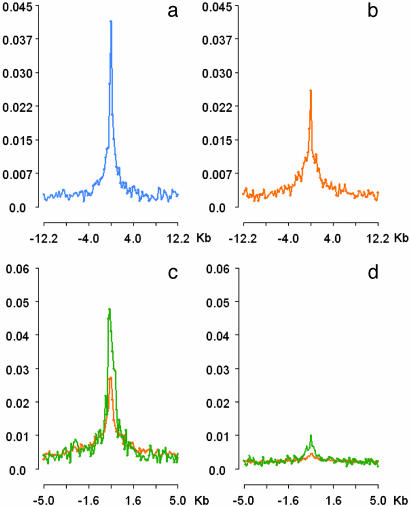
Comprehensive identification of sequences that regulate transcription is one of the major goals of genome biology. Focal alteration in chromatin structure in vivo, detectable through hypersensitivity to DNaseI and other nucleases, is the sine qua non of a diverse cast of transcriptional regulatory elements including enhancers, promoters, insulators, and locus control regions. We developed an approach for genome-scale identification of DNaseI hypersensitive sites (HSs) via isolation and cloning of in vivo DNaseI cleavage sites to create libraries of active chromatin sequences (ACSs). Here, we describe analysis of >61,000 ACSs derived from erythroid cells. We observed peaks in the density of ACSs at the transcriptional start sites of known genes at non-gene-associated CpG islands, and, to a lesser degree, at evolutionarily conserved noncoding sequences. Peaks in ACS density paralleled the distribution of DNaseI HSs. ACSs and DNaseI HSs were distributed between both expressed and nonexpressed genes, suggesting that a large proportion of genes reside within open chromatin domains. The results permit a quantitative approximation of the distribution of HSs and classical cis-regulatory sequences in the human genome.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
