

Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition
- PMID: 15075377
- PMCID: PMC420103
- DOI: 10.1091/mbc.e03-12-0879
Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition
Abstract
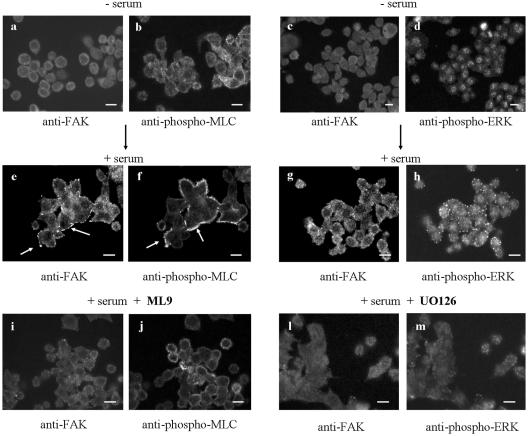
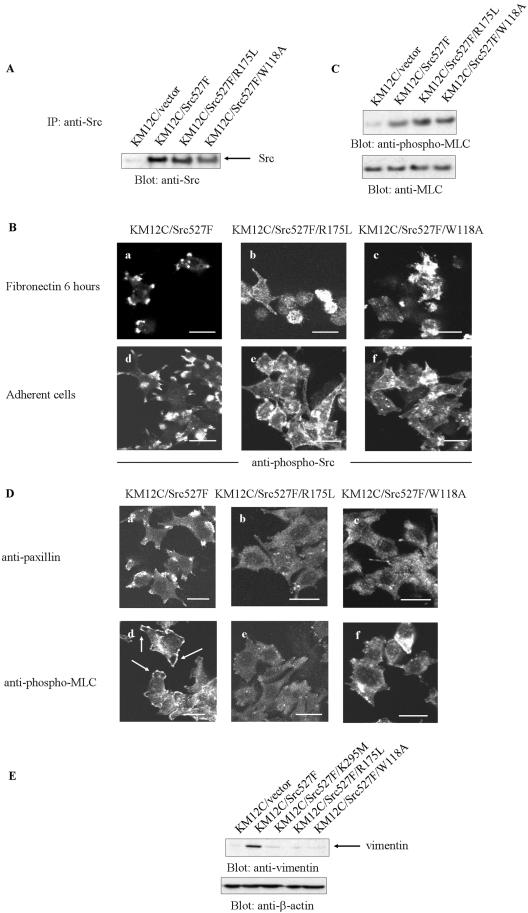
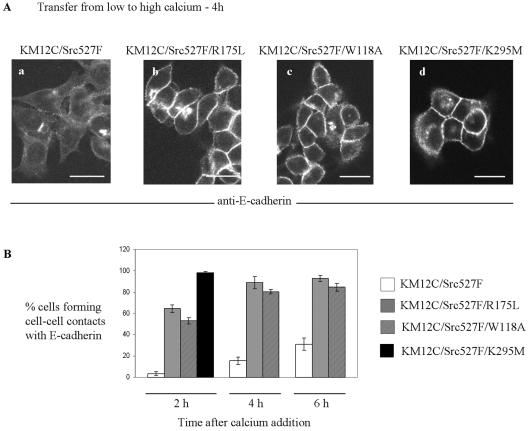
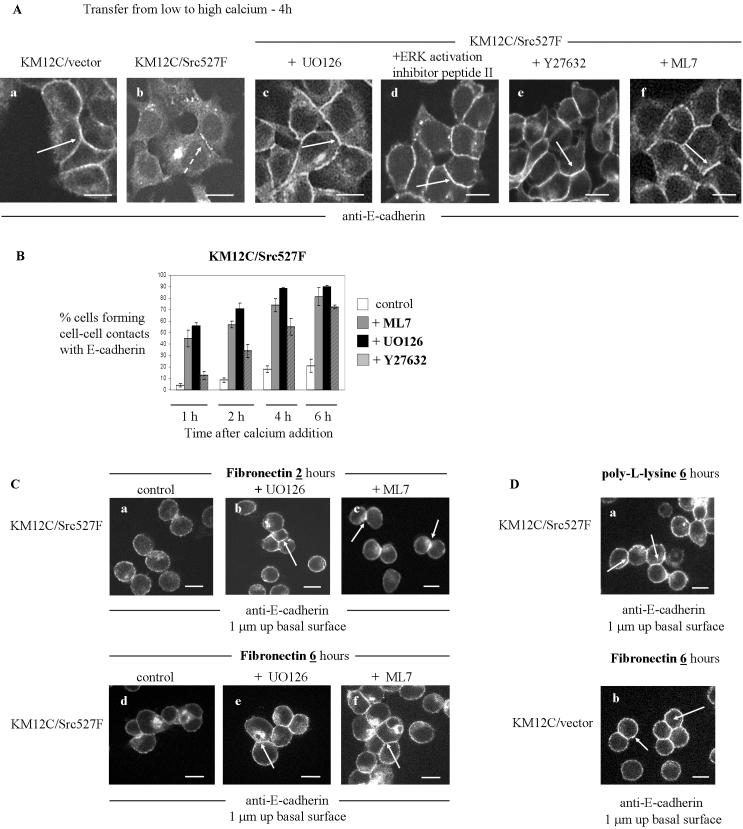
Elevated Src kinase in epithelial cancer cells induces adhesion changes that are associated with a mesenchymal-like state. We recently showed that Src induces dynamic integrin adhesions in KM12C colon cancer cells, whereas E-cadherin-dependent cell-cell contacts become disorganized. This promotes a fibroblastic-like morphology and expression of the mesenchymal marker vimentin. Furthermore, Src-induced deregulation of E-cadherin, and the associated mesenchymal transition, is dependent on integrin signaling (Avizienyte et al., Nat. Cell Biol. 2002, 4, 632-638), although the nature of downstream signals that mediate these Src- and integrin-dependent effects are unknown. Here we show that the SH2 and SH3 domains of Src mediate peripheral accumulation of phospho-myosin, leading to integrin adhesion complex assembly, whereas loss of SH2 or SH3 function restores normal regulation of E-cadherin and inhibits vimentin expression. Inhibitors of MEK, ROCK, or MLCK also suppress peripheral accumulation of phospho-myosin and Src-induced formation of integrin-dependent adhesions, whereas at the same time restoring E-cadherin redistribution to regions of cell-cell contact. Our data therefore implicate peripheral phospho-myosin activity as a point of convergence for upstream signals that regulate integrin- and E-cadherin-mediated adhesions. This further implicates spatially regulated contractile force as a determinant of epithelial cell plasticity, particularly in cancer cells that can switch between epithelial and mesenchymal-like states.
Figures


References
-
- Avizienyte, E., Wyke, A.W., Jones, R.J., McLean, G.W., Westhoff, M.A., Brunton, V.G., and Frame, M.C. (2002). Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat. Cell Biol. 4, 632-638. - PubMed
-
- Boyer, B., Bourgeois, Y., and Poupon, M.F. (2002). Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene 21, 2347-2356. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
