

HRC is a direct transcriptional target of MEF2 during cardiac, skeletal, and arterial smooth muscle development in vivo
- PMID: 15082771
- PMCID: PMC387749
- DOI: 10.1128/MCB.24.9.3757-3768.2004
HRC is a direct transcriptional target of MEF2 during cardiac, skeletal, and arterial smooth muscle development in vivo
Abstract
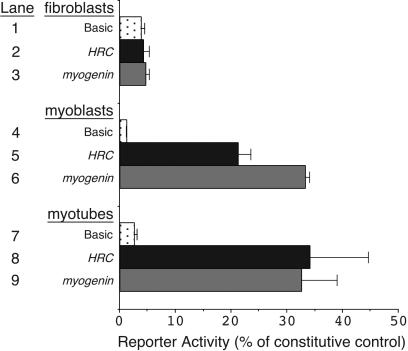
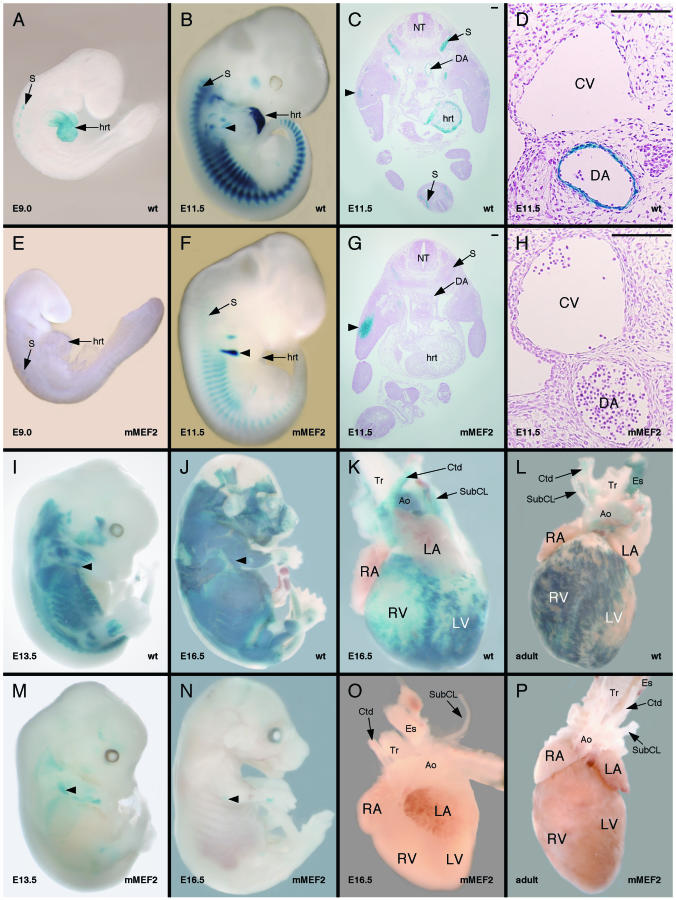
The HRC gene encodes the histidine-rich calcium-binding protein, which is found in the lumen of the junctional sarcoplasmic reticulum (SR) of cardiac and skeletal muscle and within calciosomes of arterial smooth muscle. The expression of HRC in cardiac, skeletal, and smooth muscle raises the possibility of a common transcriptional mechanism governing its expression in all three muscle cell types. In this study, we identified a transcriptional enhancer from the HRC gene that is sufficient to direct the expression of lacZ in the expression pattern of endogenous HRC in transgenic mice. The HRC enhancer contains a small, highly conserved sequence that is required for expression in all three muscle lineages. Within this conserved region is a consensus site for myocyte enhancer factor 2 (MEF2) proteins that we show is bound efficiently by MEF2 and is required for transgene expression in all three muscle lineages in vivo. Furthermore, the entire HRC enhancer sequence lacks any discernible CArG motifs, the binding site for serum response factor (SRF), and we show that the enhancer is not activated by SRF. Thus, these studies identify the HRC enhancer as the first MEF2-dependent, CArG-independent transcriptional target in smooth muscle and represent the first analysis of the transcriptional regulation of an SR gene in vivo.
Figures





References
-
- Andres, V., M. Cervera, and V. Mahdavi. 1995. Determination of the consensus binding site for MEF2 expressed in muscle and brain reveals tissue-specific sequence constraints. J. Biol. Chem. 270:23246-23249. - PubMed
-
- Baker, D. L., V. Dave, T. Reed, and M. Periasamy. 1996. Multiple Sp1 binding sites in the cardiac/slow twitch muscle sarcoplasmic reticulum Ca2+-ATPase gene promoter are required for expression in Sol8 muscle cells. J. Biol. Chem. 271:5921-5928. - PubMed
-
- Bi, W., C. J. Drake, and J. J. Schwarz. 1999. The transcription factor MEF2C-null mouse exhibits complex vascular malformations and reduced cardiac expression of angiopoietin 1 and VEGF. Dev. Biol. 211:255-267. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous