

Molecular analysis using DHPLC of cystic fibrosis: increase of the mutation detection rate among the affected population in Central Italy
- PMID: 15084222
- PMCID: PMC419352
- DOI: 10.1186/1471-2350-5-8
Molecular analysis using DHPLC of cystic fibrosis: increase of the mutation detection rate among the affected population in Central Italy
Abstract
Background: Cystic fibrosis (CF) is a multisystem disorder characterised by mutations of the CFTR gene, which encodes for an important component in the coordination of electrolyte movement across of epithelial cell membranes. Symptoms are pulmonary disease, pancreatic exocrine insufficiency, male infertility and elevated sweat concentrations. The CFTR gene has numerous mutations (>1000) and functionally important polymorphisms (>200). Early identification is important to provide appropriate therapeutic interventions, prognostic and genetic counselling and to ensure access to specialised medical services. However, molecular diagnosis by direct mutation screening has proved difficult in certain ethnic groups due to allelic heterogeneity and variable frequency of causative mutations.
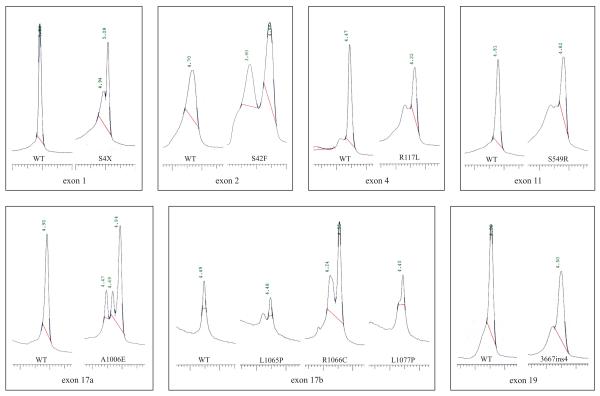
Methods: We applied a gene scanning approach using DHPLC system for analysing specifically all CFTR exons and characterise sequence variations in a subgroup of CF Italian patients from the Lazio region (Central Italy) characterised by an extensive allelic heterogeneity.
Results: We have identified a total of 36 different mutations representing 88% of the CF chromosomes. Among these are two novel CFTR mutations, including one missense (H199R) and one microdeletion (4167delCTAAGCC).
Conclusion: Using this approach, we were able to increase our standard power rate of mutation detection of about 11% (77% vs. 88%).
Figures
References
-
- Welsh MJ, Tsui LC, Boat TF, Beaudet AL. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editor. In The metabolic and molecular bases of inherited disease. 7. McGraw Hill, New York; 1995. pp. 3799–3876.
-
- Rommens JM, Iannuzzi MC, Kerem BS, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan J, Tsui LC, Collins FS. Identification of cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. - PubMed
-
- Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm M, Iannuzi MC, Collins FS, Tsui LC. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
