

Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices
- PMID: 15084665
- PMCID: PMC6729351
- DOI: 10.1523/JNEUROSCI.5539-03.2004
Sequential release of GABA by exocytosis and reversed uptake leads to neuronal swelling in simulated ischemia of hippocampal slices
Abstract
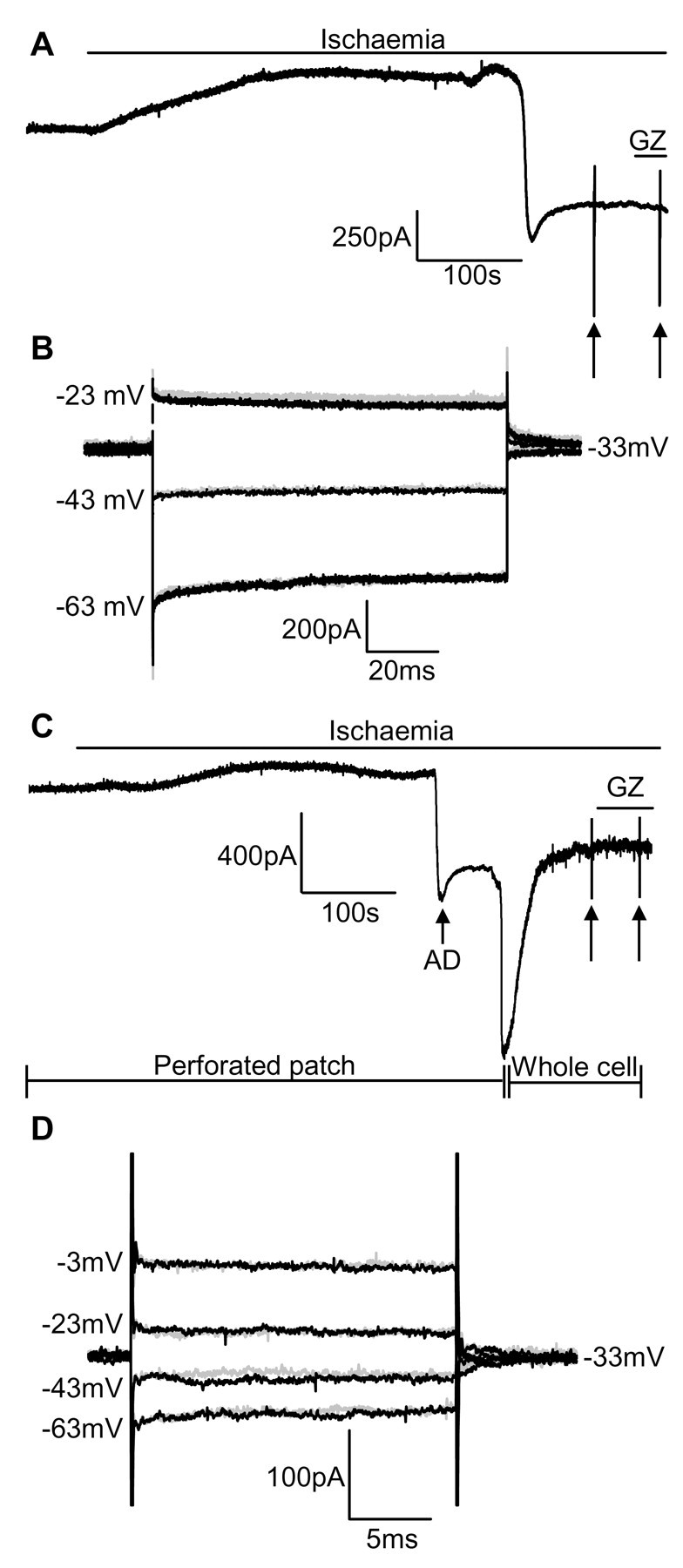
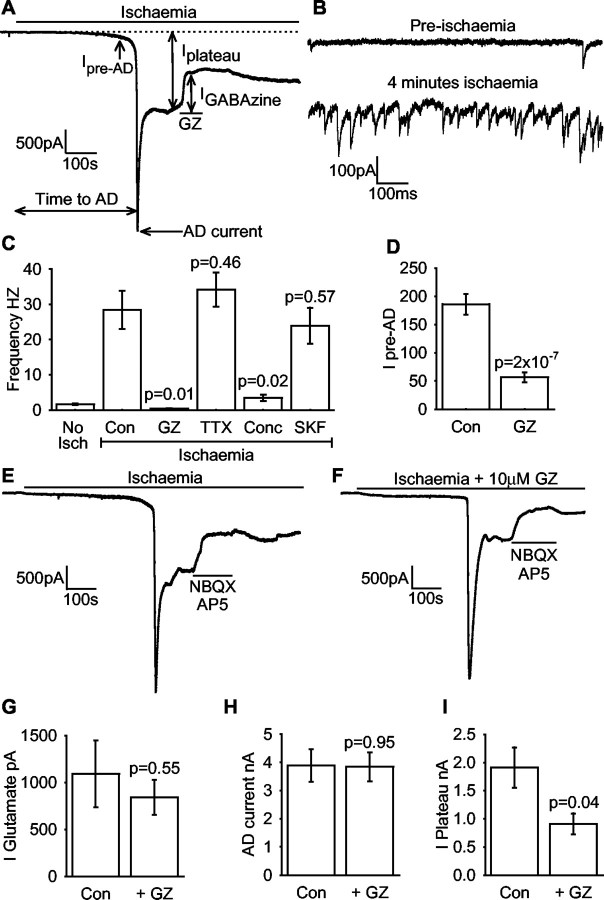
GABA release during cerebral energy deprivation (produced by anoxia or ischemia) has been suggested either to be neuroprotective, because GABA will hyperpolarize neurons and reduce release of excitotoxic glutamate, or to be neurotoxic, because activation of GABA(A) receptors facilitates Cl- entry into neurons and consequent cell swelling. We have used the GABA(A) receptors of hippocampal area CA1 pyramidal cells to sense the rise of [GABA](o) occurring in simulated ischemia. Ischemia evoked, after several minutes, a large depolarization to approximately -20 mV. Before this "anoxic depolarization," there was an increase in GABA release by exocytosis (spontaneous IPSCs). After the anoxic depolarization, there was a much larger, sustained release of GABA that was not affected by blocking action potentials, vesicular release, or the glial GABA transporter GAT-3 but was inhibited by blocking the neuronal GABA transporter GAT-1. Blocking GABA(A) receptors resulted in a more positive anoxic depolarization but decreased cell swelling at the time of the anoxic depolarization. The influence of GABA(A) receptors diminished in prolonged ischemia because glutamate release evoked by the anoxic depolarization inhibited GABA(A) receptor function by causing calcium entry through NMDA receptors. These data show that ischemia releases GABA initially by exocytosis and then by reversal of GAT-1 transporters and that the resulting Cl- influx through GABA(A) receptor channels causes potentially neurotoxic cell swelling.
Figures







References
-
- Aika Y, Ren JQ, Kosaka K, Kosaka T (1994) Quantitative analysis of GABA-like-immunoreactive and parvalbumin-containing neurons in the CA1 region of the rat hippocampus using a stereological method, the disector. Exp Brain Res 99: 267–276. - PubMed
-
- Akaike N (1996) Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Prog Biophys Mol Biol 65: 251–264. - PubMed
-
- Alicke B, Schwartz-Bloom RB (1995) Rapid down-regulation of GABAA receptors in the gerbil hippocampus following transient cerebral ischaemia. J Neurochem 65: 2808–2811. - PubMed
-
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P (1995) Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15: 961–973. - PubMed
-
- Attwell D, Barbour B, Szatkowski M (1993) Nonvesicular release of neurotransmitter. Neuron 11: 401–407. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous