

Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway
- PMID: 15102801
- PMCID: PMC387848
- DOI: 10.1128/IAI.72.5.2889-2898.2004
Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway
Abstract
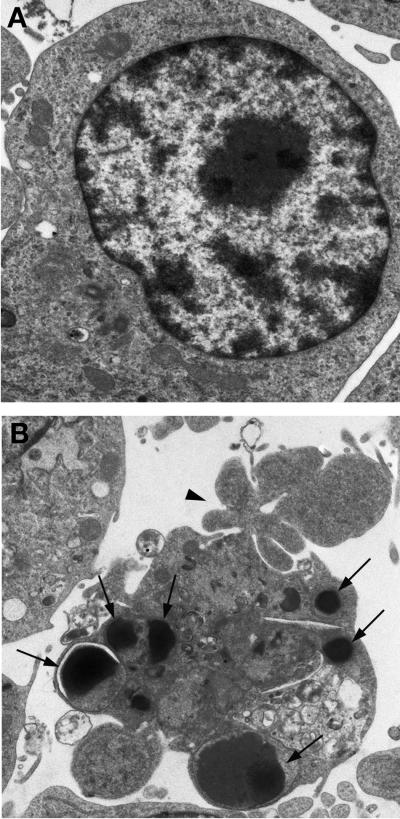
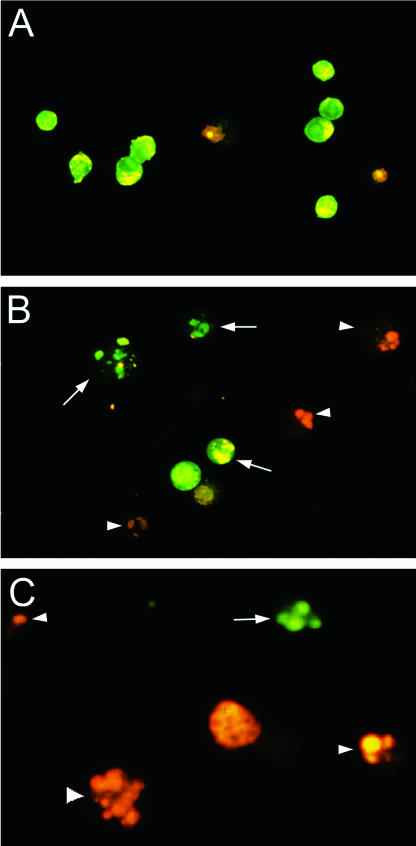
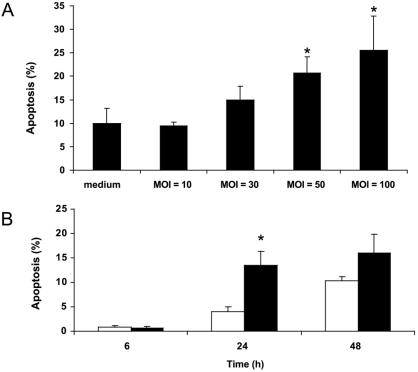
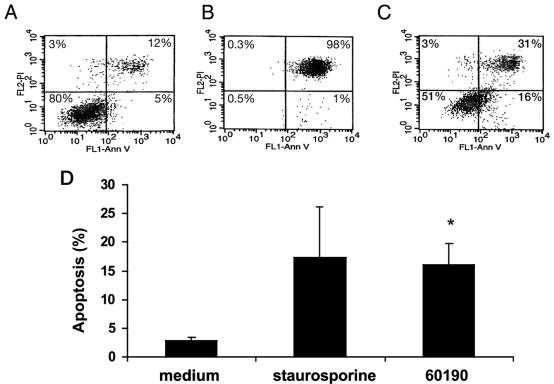
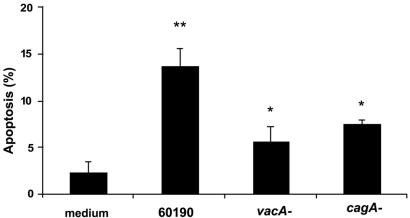
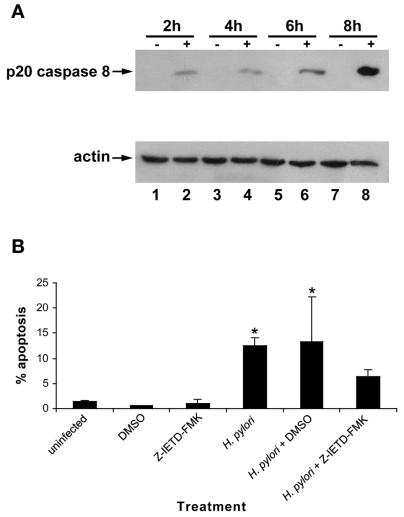
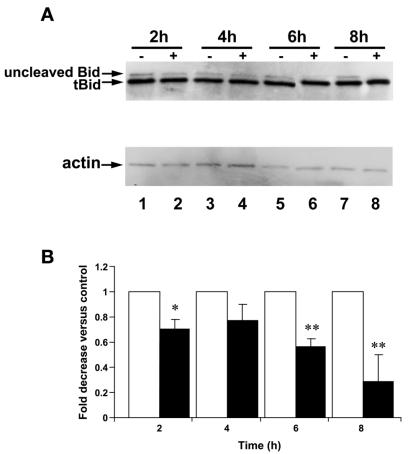
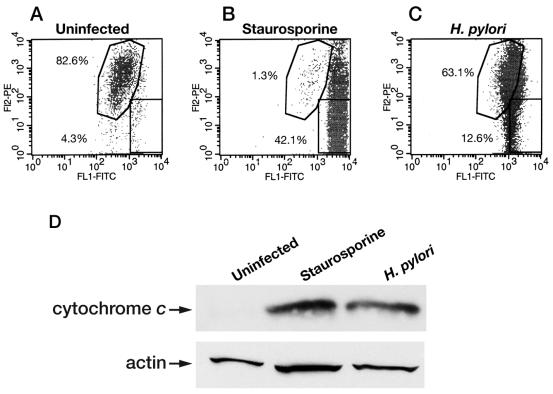
Helicobacter pylori is a gastric bacterial pathogen that evades host immune responses in vivo and is associated with the development of gastritis, peptic ulcer disease, and gastric cancers. Induction of macrophage apoptosis is a method employed by multiple pathogens to escape host immune responses. Therefore, we hypothesized that H. pylori induces apoptosis of infected macrophages. RAW 264.7 cells were infected with H. pylori strain 60190, and apoptosis was assessed. Transmission electron microscopy and fluorescence microscopy showed that infected macrophages displayed morphological features characteristic of apoptosis. Quantification by acridine orange-ethidium bromide fluorescent-dye staining showed that apoptosis was dose and time dependent, and apoptosis was further confirmed by increased binding of annexin V-fluorescein isothiocyanate (FITC) to externalized phosphatidylserine of infected but not of control macrophages. Macrophages infected with isogenic mutants of H. pylori strain 60190 deficient in either cagA or vacA induced significantly less apoptosis than the parental strain, as assessed by increased binding of annexin V-FITC. Western blot analysis of whole-cell protein lysates revealed that infection with strain 60190 induced a time-dependent increase in cleavage of procaspase 8 and disappearance of full-length Bid compared with uninfected cells. Furthermore, pharmacological inhibition of caspase 8 caused a decrease in levels of apoptosis. Finally, infection caused a time-dependent increase in mitochondrial-membrane permeability and release of cytochrome c into the cytosol. These results suggest that H. pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Elimination of this key immunomodulatory cell may represent a mechanism employed by the bacterium to evade host immune responses.
Figures
Similar articles
-
Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization.J Biol Chem. 2004 Sep 17;279(38):40161-73. doi: 10.1074/jbc.M401370200. Epub 2004 Jul 9. J Biol Chem. 2004. PMID: 15247269
-
Helicobacter pylori Secreted Protein HP1286 Triggers Apoptosis in Macrophages via TNF-Independent and ERK MAPK-Dependent Pathways.Front Cell Infect Microbiol. 2017 Feb 28;7:58. doi: 10.3389/fcimb.2017.00058. eCollection 2017. Front Cell Infect Microbiol. 2017. PMID: 28293545 Free PMC article.
-
Helicobacter pylori inhibits the cleavage of TRAF1 via a CagA-dependent mechanism.World J Gastroenterol. 2016 Dec 28;22(48):10566-10574. doi: 10.3748/wjg.v22.i48.10566. World J Gastroenterol. 2016. PMID: 28082808 Free PMC article.
-
Influence of Helicobacter pylori virulence factors CagA and VacA on pathogenesis of gastrointestinal disorders.Microb Pathog. 2018 Apr;117:43-48. doi: 10.1016/j.micpath.2018.02.016. Epub 2018 Feb 9. Microb Pathog. 2018. PMID: 29432909 Review.
-
Macrophage biology in the pathogenesis of Helicobacter pylori infection.Crit Rev Microbiol. 2025 May;51(3):399-416. doi: 10.1080/1040841X.2024.2366944. Epub 2024 Jul 31. Crit Rev Microbiol. 2025. PMID: 39086061 Review.
Cited by
-
Fucosyltransferase-4 and Oligosaccharide Lewis Y Antigen as potentially Correlative Biomarkers of Helicobacter pylori CagA Associated Gastric Cancer.Pathol Oncol Res. 2017 Jan;23(1):173-179. doi: 10.1007/s12253-016-0122-1. Epub 2016 Oct 18. Pathol Oncol Res. 2017. PMID: 27757838
-
Suppression of cell division-associated genes by Helicobacter pylori attenuates proliferation of RAW264.7 monocytic macrophage cells.Sci Rep. 2015 Jun 16;5:11046. doi: 10.1038/srep11046. Sci Rep. 2015. PMID: 26078204 Free PMC article.
-
Prolonged classical NF-kappaB activation prevents autophagy upon E. coli stimulation in vitro: a potential resolving mechanism of inflammation.Mediators Inflamm. 2008;2008:725854. doi: 10.1155/2008/725854. Mediators Inflamm. 2008. PMID: 18566685 Free PMC article.
-
Helicobacter pylori infection and Parkinson's Disease: etiology, pathogenesis and levodopa bioavailability.Immun Ageing. 2024 Jan 2;21(1):1. doi: 10.1186/s12979-023-00404-1. Immun Ageing. 2024. PMID: 38166953 Free PMC article. Review.
-
Restoration of mitochondrial structure and function within Helicobacter pylori VacA intoxicated cells.Adv Microbiol. 2023 Aug;13(8):399-419. doi: 10.4236/aim.2023.138026. Adv Microbiol. 2023. PMID: 37654621 Free PMC article.
References
-
- Allen, L. A. 1999. Intracellular niches for extracellular bacteria: lessons from Helicobacter pylori. J. Leukoc. Biol. 66:753-756. - PubMed
-
- Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signalling and modulation. Science 281:1305-1308. - PubMed
-
- Ashktorab, H., M. Neapolitano, C. Bomma, C. Allen, A. Ahmed, A. Dubois, T. Naab, and D. T. Smoot. 2002. In vivo and in vitro activation of caspase-8 and -3 associated with Helicobacter pylori infection. Microb. Infect. 4:713-722. - PubMed
-
- Backert, S., S. Moese, M. Selbach, V. Brinkmann, and T. F. Meyer. 2001. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol. Microbiol. 42:631-644. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
