

Integrative annotation of 21,037 human genes validated by full-length cDNA clones
- PMID: 15103394
- PMCID: PMC393292
- DOI: 10.1371/journal.pbio.0020162
Integrative annotation of 21,037 human genes validated by full-length cDNA clones
Erratum in
- PLoS Biol. 2004 Jul;2(7):e256
Abstract
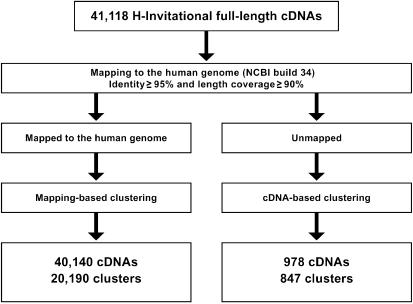
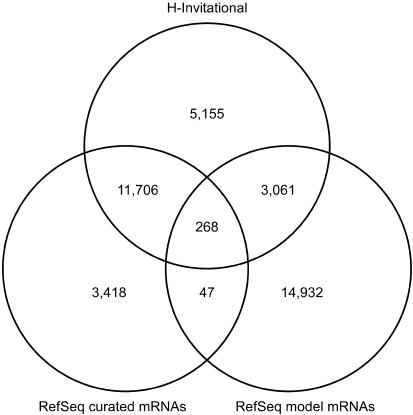
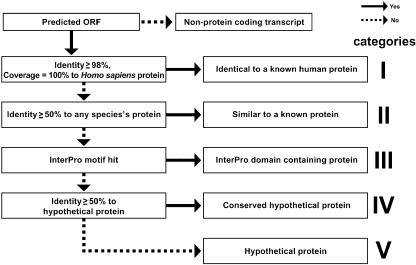
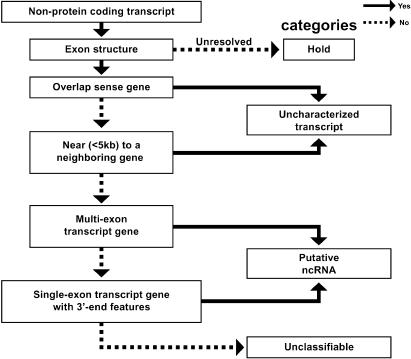
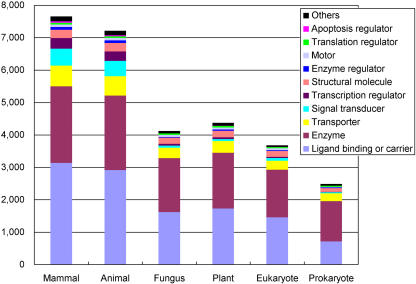
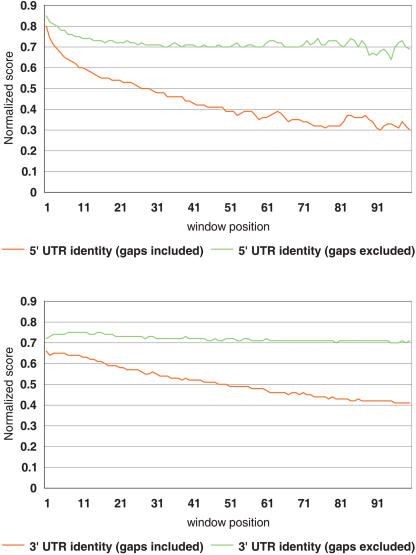
The human genome sequence defines our inherent biological potential; the realization of the biology encoded therein requires knowledge of the function of each gene. Currently, our knowledge in this area is still limited. Several lines of investigation have been used to elucidate the structure and function of the genes in the human genome. Even so, gene prediction remains a difficult task, as the varieties of transcripts of a gene may vary to a great extent. We thus performed an exhaustive integrative characterization of 41,118 full-length cDNAs that capture the gene transcripts as complete functional cassettes, providing an unequivocal report of structural and functional diversity at the gene level. Our international collaboration has validated 21,037 human gene candidates by analysis of high-quality full-length cDNA clones through curation using unified criteria. This led to the identification of 5,155 new gene candidates. It also manifested the most reliable way to control the quality of the cDNA clones. We have developed a human gene database, called the H-Invitational Database (H-InvDB; http://www.h-invitational.jp/). It provides the following: integrative annotation of human genes, description of gene structures, details of novel alternative splicing isoforms, non-protein-coding RNAs, functional domains, subcellular localizations, metabolic pathways, predictions of protein three-dimensional structure, mapping of known single nucleotide polymorphisms (SNPs), identification of polymorphic microsatellite repeats within human genes, and comparative results with mouse full-length cDNAs. The H-InvDB analysis has shown that up to 4% of the human genome sequence (National Center for Biotechnology Information build 34 assembly) may contain misassembled or missing regions. We found that 6.5% of the human gene candidates (1,377 loci) did not have a good protein-coding open reading frame, of which 296 loci are strong candidates for non-protein-coding RNA genes. In addition, among 72,027 uniquely mapped SNPs and insertions/deletions localized within human genes, 13,215 nonsynonymous SNPs, 315 nonsense SNPs, and 452 indels occurred in coding regions. Together with 25 polymorphic microsatellite repeats present in coding regions, they may alter protein structure, causing phenotypic effects or resulting in disease. The H-InvDB platform represents a substantial contribution to resources needed for the exploration of human biology and pathology.
Conflict of interest statement
The authors have declared that no conflicts of interest exist.
Figures
References
-
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science. 1991;252:1651–1656. - PubMed
-
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. The genome sequence of Drosophila melanogaster . Science. 2000;287:2185–2195. - PubMed
-
- [AGI] Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
