

Mutations in the human TBX4 gene cause small patella syndrome
- PMID: 15106123
- PMCID: PMC1182087
- DOI: 10.1086/421331
Mutations in the human TBX4 gene cause small patella syndrome
Abstract
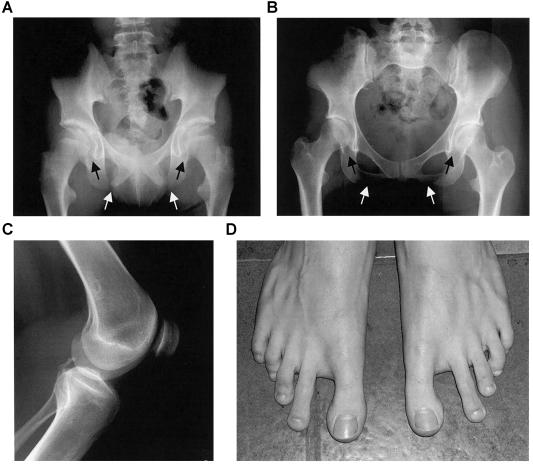
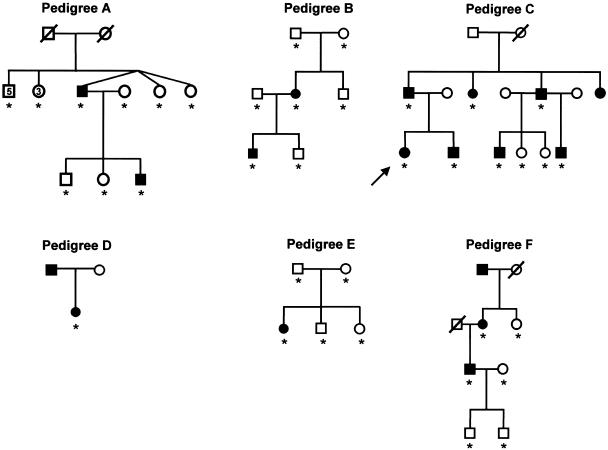
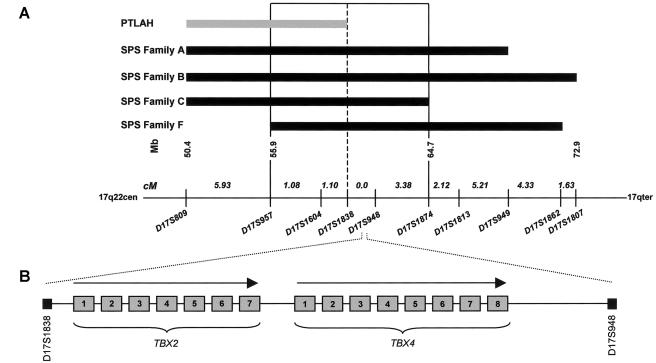
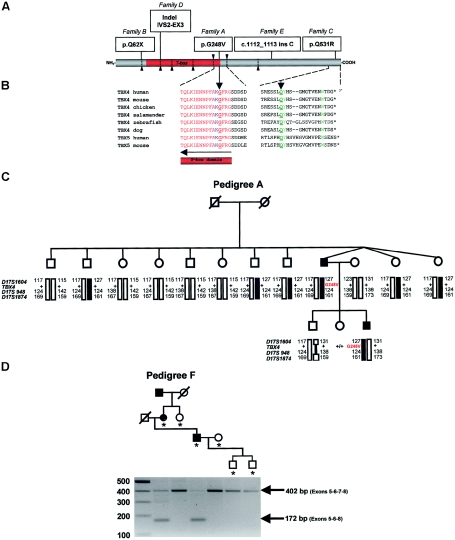
Small patella syndrome (SPS) is an autosomal-dominant skeletal dysplasia characterized by patellar aplasia or hypoplasia and by anomalies of the pelvis and feet, including disrupted ossification of the ischia and inferior pubic rami. We identified an SPS critical region of 5.6 cM on chromosome 17q22 by haplotype analysis. Putative loss-of-function mutations were found in a positional gene encoding T-box protein 4 (TBX4) in six families with SPS. TBX4 encodes a transcription factor with a strongly conserved DNA-binding T-box domain that is known to play a crucial role in lower limb development in chickens and mice. The present identification of heterozygous TBX4 mutations in SPS patients, together with the similar skeletal phenotype of animals lacking Tbx4, establish the importance of TBX4 in the developmental pathways of the lower limbs and the pelvis in humans.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (for TBX4 [H. sapiens] mRNA [accession number AF188703] and for TBX2 [H. sapiens] mRNA [accession number U28049])
-
- NCBI BLAST, http://www.ncbi.nlm.gov/BLAST/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
-
- Protein Data Bank, http://www.rcsb.org/pdb/ (for TBX3 [accession number 1H6F])
-
- UCSC Genome Bioinformatics, http://genome.cse.ucsc.edu/ (for Human Genome Browser)
References
-
- Bernhang AM, Levine SA (1973) Familial absence of the patella. J Bone Joint Surg Am 55:1088–1090 - PubMed
-
- Bamshad M, Lin R, Law Dj, Watkins WC, Krakowiak PA, Moore ME, Fransceschini P, Lala R, Holmes LB, Gebuhr TC, Bruneau GB, Schinzel A, Seidman JG, Seidman CE, Jorde LB (1997) Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet 16:311–315 - PubMed
-
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill T, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE (1997) Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 15:30–35 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
