

Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data
- PMID: 15113405
- PMCID: PMC407845
- DOI: 10.1186/1471-2105-5-31
Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data
Abstract
Background: Functional genomics studies are yielding information about regulatory processes in the cell at an unprecedented scale. In the yeast S. cerevisiae, DNA microarrays have not only been used to measure the mRNA abundance for all genes under a variety of conditions but also to determine the occupancy of all promoter regions by a large number of transcription factors. The challenge is to extract useful information about the global regulatory network from these data.
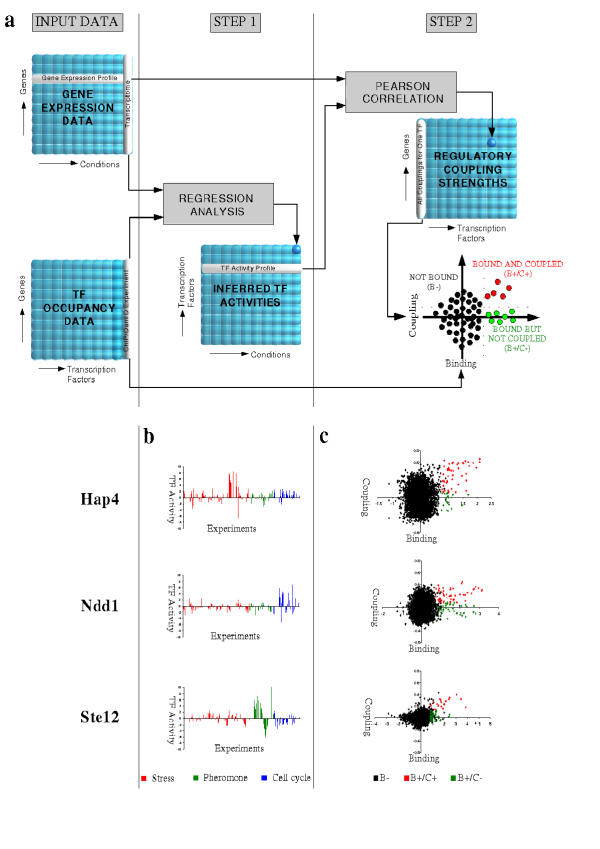
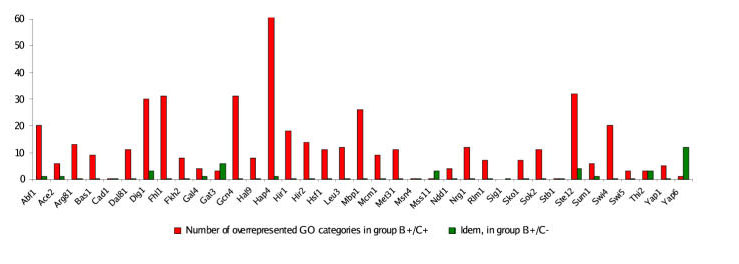
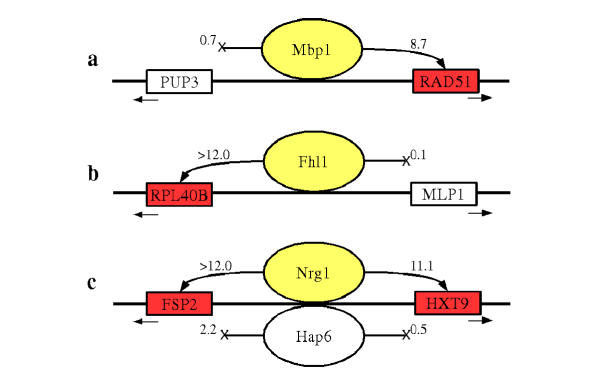
Results: We present MA-Networker, an algorithm that combines microarray data for mRNA expression and transcription factor occupancy to define the regulatory network of the cell. Multivariate regression analysis is used to infer the activity of each transcription factor, and the correlation across different conditions between this activity and the mRNA expression of a gene is interpreted as regulatory coupling strength. Applying our method to S. cerevisiae, we find that, on average, 58% of the genes whose promoter region is bound by a transcription factor are true regulatory targets. These results are validated by an analysis of enrichment for functional annotation, response for transcription factor deletion, and over-representation of cis-regulatory motifs. We are able to assign directionality to transcription factors that control divergently transcribed genes sharing the same promoter region. Finally, we identify an intrinsic limitation of transcription factor deletion experiments related to the combinatorial nature of transcriptional control, to which our approach provides an alternative.
Conclusion: Our reliable classification of ChIP positives into functional and non-functional TF targets based on their expression pattern across a wide range of conditions provides a starting point for identifying the unknown sequence features in non-coding DNA that directly or indirectly determine the context dependence of transcription factor action. Complete analysis results are available for browsing or download at http://bussemaker.bio.columbia.edu/papers/MA-Networker/.
Figures
References
-
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. - PubMed
-
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
