

The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome
- PMID: 15138899
- PMCID: PMC1182011
- DOI: 10.1086/421846
The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome
Abstract
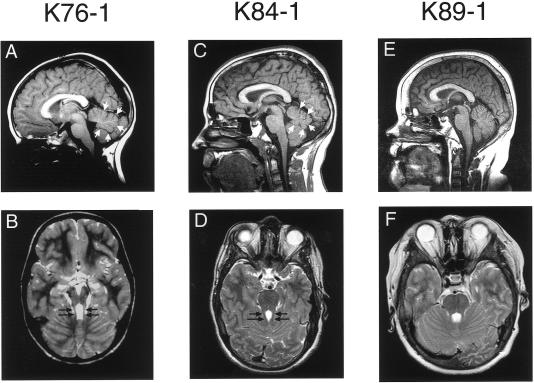
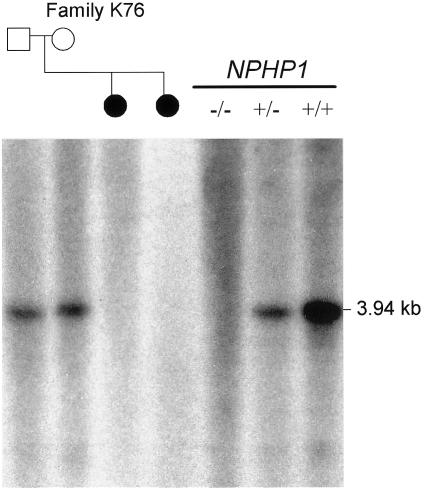
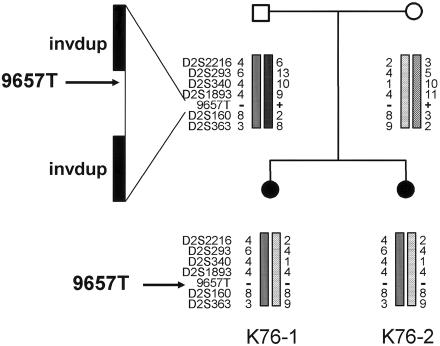
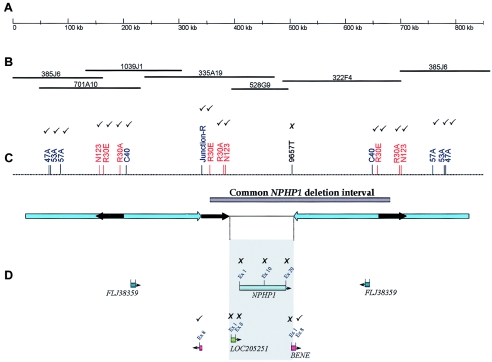
Joubert syndrome (JS) is an autosomal recessive multisystem disease characterized by cerebellar vermis hypoplasia with prominent superior cerebellar peduncles (the "molar tooth sign" [MTS] on axial magnetic resonance imaging), mental retardation, hypotonia, irregular breathing pattern, and eye-movement abnormalities. Some individuals with JS have retinal dystrophy and/or progressive renal failure characterized by nephronophthisis (NPHP). Thus far, no mutations in the known NPHP genes, particularly the homozygous deletion of NPHP1 at 2q13, have been identified in subjects with JS. A cohort of 25 subjects with JS and either renal and/or retinal complications and 2 subjects with only juvenile NPHP were screened for mutations in the NPHP1 gene by standard methods. Two siblings affected with a mild form of JS were found to have a homozygous deletion of the NPHP1 gene identical, by mapping, to that in subjects with NPHP alone. A control subject with NPHP and with a homozygous NPHP1 deletion was also identified, retrospectively, as having a mild MTS and borderline intelligence. The NPHP1 deletion represents the first molecular defect associated with JS in a subset of mildly affected subjects. Cerebellar malformations consistent with the MTS may be relatively common in patients with juvenile NPHP without classic symptoms of JS.
Figures
References
Electronic-Database Information
-
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for SNP numbers rs2271245, rs2271244, rs3817140, rs1509417, and ss22970415)
-
- GeneReviews at GeneTests-GeneClinics, http://www.geneclinics.org or http://www.genetests.org
-
- Joubert Syndrome Foundation, http://www.joubertsyndrome.org
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
-
- UCSC human genome browser, http://genome.ucsc.edu/cgi-bin/hgGateway?org=human
References
-
- Antignac C, Kleinknecht C, Habib R (1998) Nephronophthisis. In: Cameron D, Davison AM, Cameron JS, Grunfeld J-P, Kerr DNS, Ritz E, Winearls G (eds) Clinical nephrology. Oxford University Press, Oxford and New York, pp 2417–2426
-
- Bennett CL, Meuleman J, Chance PF, Glass IA (2003) Clinical and genetic aspects of the Joubert syndrome: a disorder characterised by cerebellar vermian hypoplasia and accompanying brainstem malformations. Curr Genomics 4:123–129
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
