

Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs
- PMID: 15140833
- PMCID: PMC419793
- DOI: 10.1101/gr.1917404
Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs
Abstract
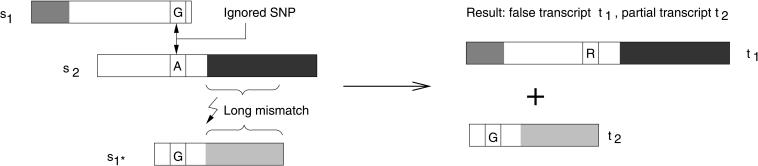
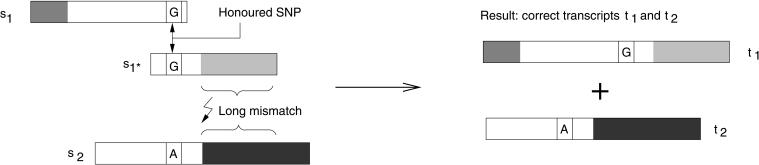
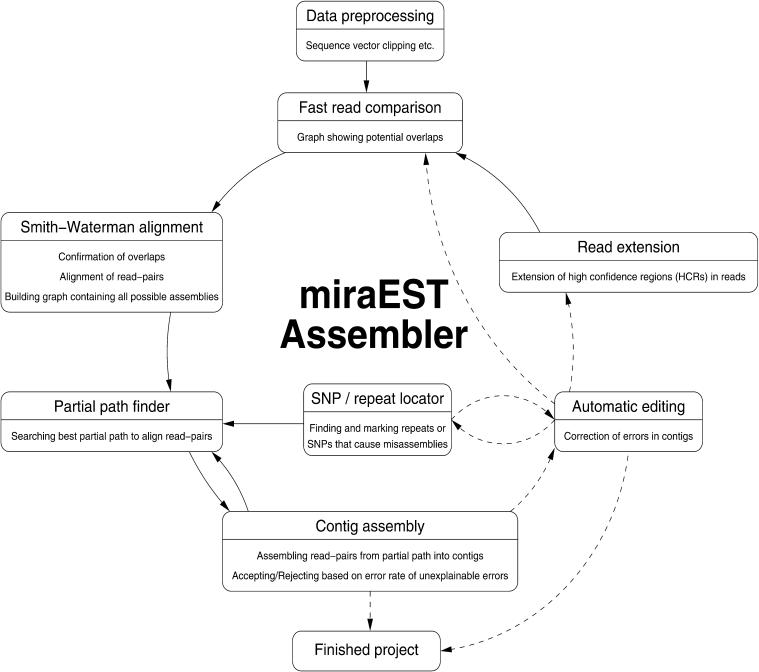
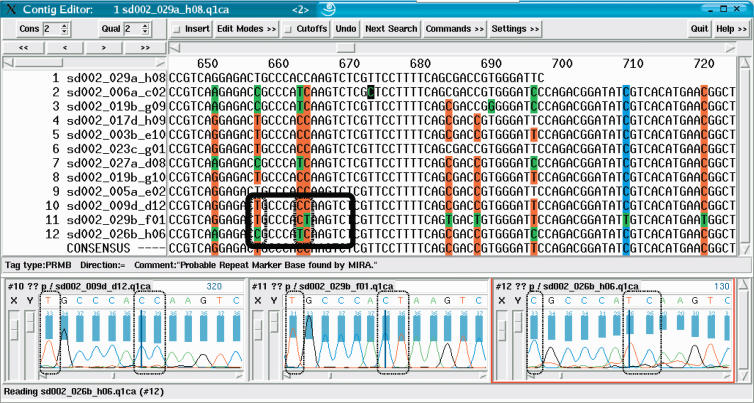
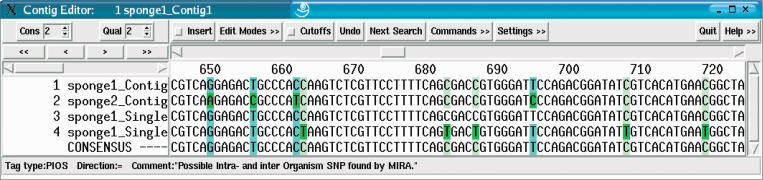
We present an EST sequence assembler that specializes in reconstruction of pristine mRNA transcripts, while at the same time detecting and classifying single nucleotide polymorphisms (SNPs) occuring in different variations thereof. The assembler uses iterative multipass strategies centered on high-confidence regions within sequences and has a fallback strategy for using low-confidence regions when needed. It features special functions to assemble high numbers of highly similar sequences without prior masking, an automatic editor that edits and analyzes alignments by inspecting the underlying traces, and detection and classification of sequence properties like SNPs with a high specificity and a sensitivity down to one mutation per sequence. In addition, it includes possibilities to use incorrectly preprocessed sequences, routines to make use of additional sequencing information such as base-error probabilities, template insert sizes, strain information, etc., and functions to detect and resolve possible misassemblies. The assembler is routinely used for such various tasks as mutation detection in different cell types, similarity analysis of transcripts between organisms, and pristine assembly of sequences from various sources for oligo design in clinical microarray experiments.
Copyright 2004 Cold Spring Harbor Laboratory Press
Figures
References
-
- Allex, C.F., Baldwin, S.F., Shavlik, J.W., and Blattner, F.R. 1996. Improving the quality of automatic DNA sequence assembly using fluorescent trace-data classifications. Intell. Systems Mol. Biol. 4: 3–14. - PubMed
-
- Arslan, A.N., Egecioglu, O., and Pevzner, P.A. 2001. A new approach to sequence comparison: Normalized sequence alignment. Bioinformatics 17: 327–337. - PubMed
-
- Baeza-Yates, R.A. and Gonnet, G.H. 1992. A new approach to text searching. Commun. of the Assoc. for Comp. Mach. 35: 74–82.
-
- Barker, G., Batley, J., O'Sullivan, H., Edwards, K.J., and Edwards, D. 2003. Redundancy based detection of sequence polymorphisms in expressed sequence tag data using autoSNP. Bioinformatics 19: 421–422. - PubMed
-
- Bonfield, J.K. and Staden, R. 1996. Experiment files and their application during large-scale sequencing projects. DNA Seq. 6: 109–117. - PubMed
WEB SITE REFERENCES
-
- http://www.chevreux.org/projects_mira.html; homepage of the MIRA V2 assembly system.
-
- http://www.dkfz.de/mbp-ased/; homepage of the MIRA V1 assembly system and EdIt automatic editor.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials