

Human immunodeficiency virus type 1 (HIV-1) integrase: resistance to diketo acid integrase inhibitors impairs HIV-1 replication and integration and confers cross-resistance to L-chicoric acid
- PMID: 15140981
- PMCID: PMC415810
- DOI: 10.1128/JVI.78.11.5835-5847.2004
Human immunodeficiency virus type 1 (HIV-1) integrase: resistance to diketo acid integrase inhibitors impairs HIV-1 replication and integration and confers cross-resistance to L-chicoric acid
Abstract
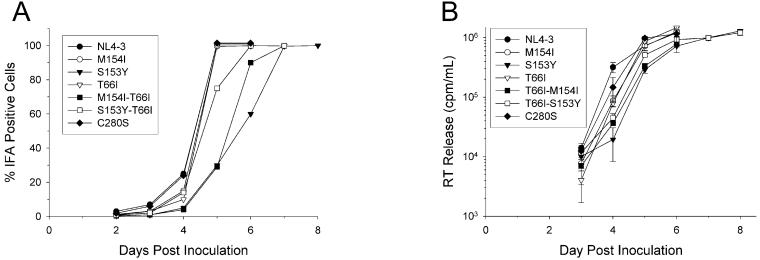
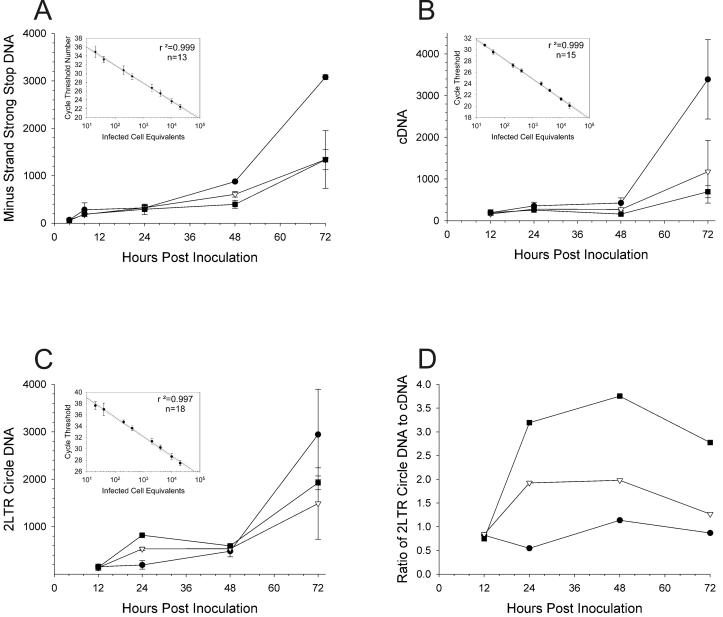
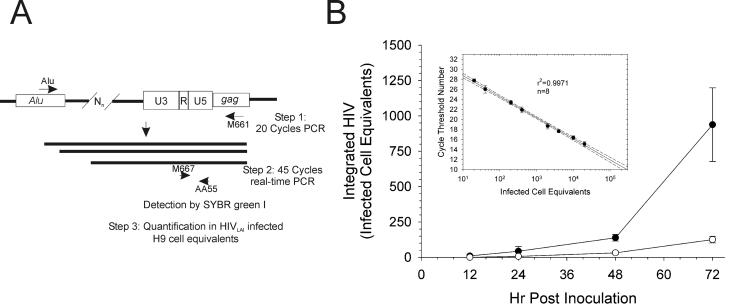
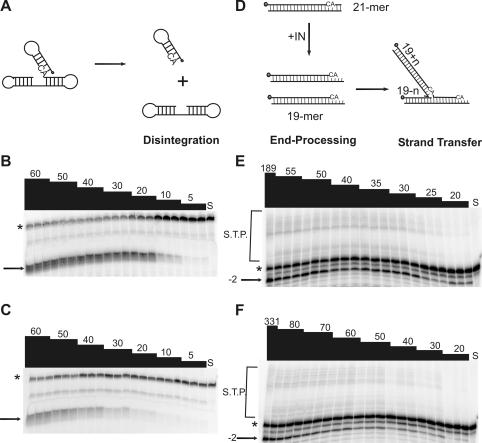
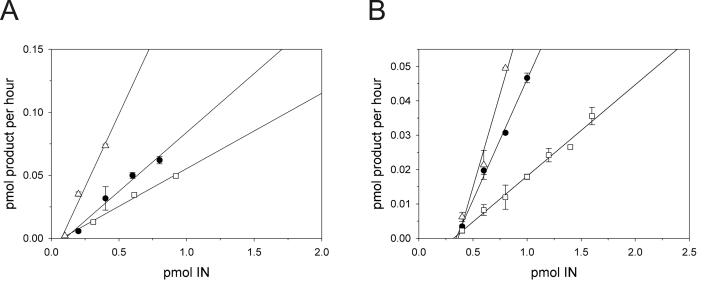
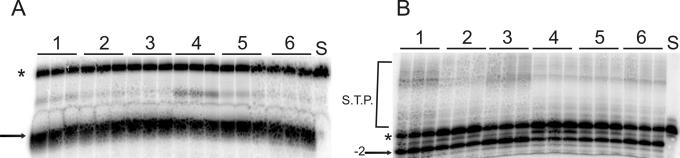
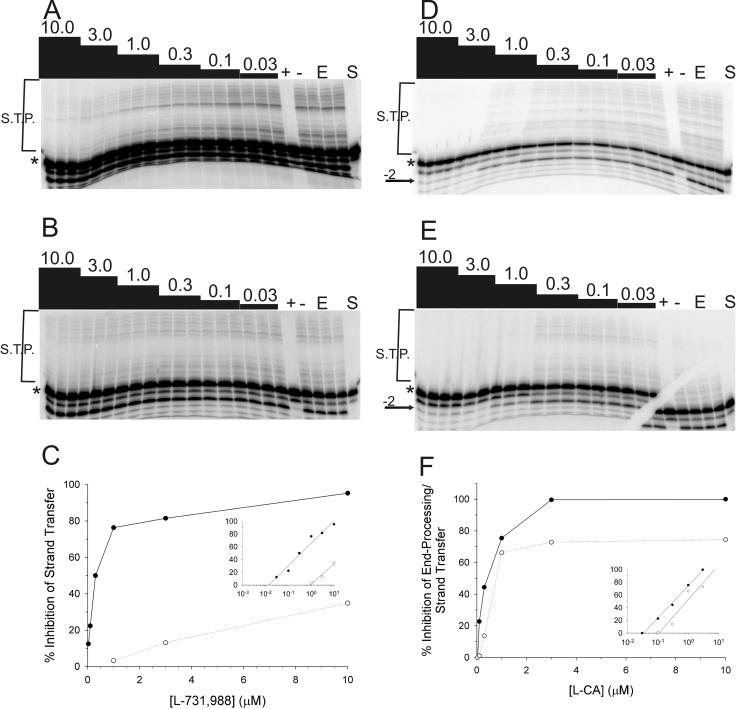
The diketo acids are potent inhibitors of human immunodeficiency virus (HIV) integrase (IN). Mutations in IN, T66I, S153Y, and M154I, as well as T66I-S153Y and T66I-M154I double mutations, confer resistance to diketo acids (D. J. Hazuda et al., Science 287:646-650, 2000). The effects of these IN mutations on viral replication, enzymatic activity, and susceptibility to other HIV inhibitors are reported herein. By immunofluorescence assay and real-time PCR, all mutant viruses demonstrated a modest delay in viral spread compared to that of reference HIV. These viruses also showed a statistically significant defect in integration without defects in reverse transcription. Recombinant IN containing S153Y, T66I, and M154I-T66I mutations had an approximately twofold decrease in both disintegration and 3'-end-processing-strand transfer activities in vitro. In contrast, IN containing M154I demonstrated a greater than twofold increase in specific activity in both reactions. All mutant HIVs were resistant to l-chicoric acid, a dicaffeoyltartaric acid IN inhibitor, both in tissue culture and in biochemical assays, yet remained susceptible to the reverse transcriptase inhibitors zidovudine and nevirapine. Thus, IN mutations conferring resistance to the diketo acids can yield integration defects, attenuated catalysis in vitro, and cross-resistance to l-chicoric acid.
Figures
Similar articles
-
Development of resistance against diketo derivatives of human immunodeficiency virus type 1 by progressive accumulation of integrase mutations.J Virol. 2003 Nov;77(21):11459-70. doi: 10.1128/jvi.77.21.11459-11470.2003. J Virol. 2003. PMID: 14557631 Free PMC article.
-
Human immunodeficiency virus type-1 integrase containing a glycine to serine mutation at position 140 is attenuated for catalysis and resistant to integrase inhibitors.Virology. 2003 Feb 1;306(1):147-61. doi: 10.1016/s0042-6822(02)00042-9. Virology. 2003. PMID: 12620807
-
Design, synthesis, and biological evaluation of novel hybrid dicaffeoyltartaric/diketo acid and tetrazole-substituted L-chicoric acid analogue inhibitors of human immunodeficiency virus type 1 integrase.J Med Chem. 2010 Nov 25;53(22):8161-75. doi: 10.1021/jm1010594. Epub 2010 Oct 26. J Med Chem. 2010. PMID: 20977258
-
Characterization and structural analysis of HIV-1 integrase conservation.AIDS Rev. 2009 Jan-Mar;11(1):17-29. AIDS Rev. 2009. PMID: 19290031 Review.
-
In search of authentic inhibitors of HIV-1 integration.Antivir Chem Chemother. 2002 Jan;13(1):1-15. doi: 10.1177/095632020201300101. Antivir Chem Chemother. 2002. PMID: 12180645 Review.
Cited by
-
Chicoric Acid Ameliorates Nonalcoholic Fatty Liver Disease via the AMPK/Nrf2/NFκB Signaling Pathway and Restores Gut Microbiota in High-Fat-Diet-Fed Mice.Oxid Med Cell Longev. 2020 Nov 3;2020:9734560. doi: 10.1155/2020/9734560. eCollection 2020. Oxid Med Cell Longev. 2020. PMID: 33204402 Free PMC article.
-
Comparison of multiple molecular dynamics trajectories calculated for the drug-resistant HIV-1 integrase T66I/M154I catalytic domain.Biophys J. 2005 May;88(5):3072-82. doi: 10.1529/biophysj.104.050286. Epub 2005 Mar 11. Biophys J. 2005. PMID: 15764656 Free PMC article.
-
Molecular dynamics approaches estimate the binding energy of HIV-1 integrase inhibitors and correlate with in vitro activity.Antimicrob Agents Chemother. 2012 Jan;56(1):411-9. doi: 10.1128/AAC.05292-11. Epub 2011 Oct 28. Antimicrob Agents Chemother. 2012. PMID: 22037850 Free PMC article.
-
Defining the DNA substrate binding sites on HIV-1 integrase.J Mol Biol. 2009 Jan 16;385(2):568-79. doi: 10.1016/j.jmb.2008.10.083. Epub 2008 Nov 7. J Mol Biol. 2009. PMID: 19014951 Free PMC article.
-
Insertions in the human immunodeficiency virus type 1 protease and reverse transcriptase genes: clinical impact and molecular mechanisms.Antimicrob Agents Chemother. 2005 Jul;49(7):2575-82. doi: 10.1128/AAC.49.7.2575-2582.2005. Antimicrob Agents Chemother. 2005. PMID: 15980322 Free PMC article. Review. No abstract available.
References
-
- Ansari-Lari, M. A., L. A. Donehower, and R. A. Gibbs. 1995. Analysis of human immunodeficiency virus type 1 integrase mutants. Virology 211:332-335. - PubMed
-
- Beale, K., and W. E. Robinson, Jr. 2000. Combinations of reverse transcriptase, protease, and integrase inhibitors can be synergistic in vitro against drug-sensitive and RT inhibitor-resistant molecular clones of HIV-1. Antivir. Res. 46:223-232. - PubMed
-
- Buolamwini, J. K., and H. Assefa. 2002. CoMFA and CoMSIA 3D QSAR and docking studies on conformationally-restrained cinnamoyl HIV-1 integrase inhibitors: exploration of a binding mode at the active site. J. Med. Chem. 45:841-852. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
