

Localization and dynamics of small circular DNA in live mammalian nuclei
- PMID: 15141035
- PMCID: PMC419472
- DOI: 10.1093/nar/gkh587
Localization and dynamics of small circular DNA in live mammalian nuclei
Abstract
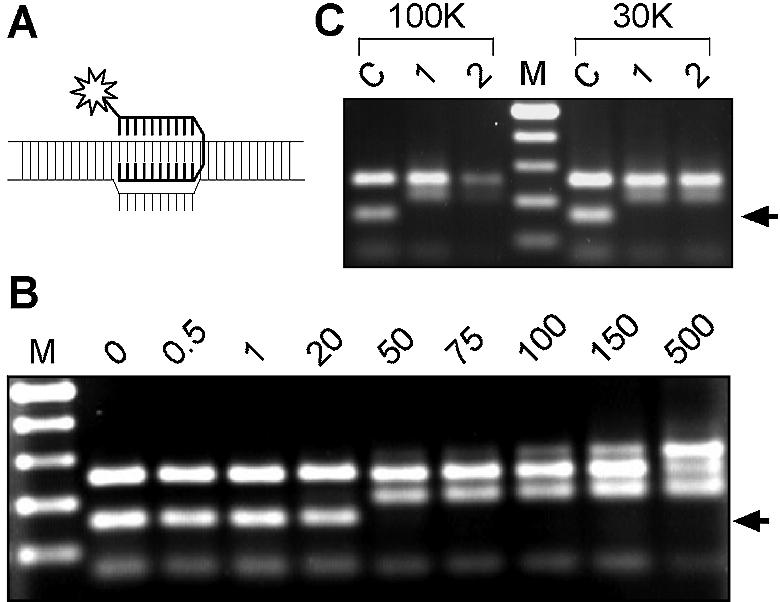
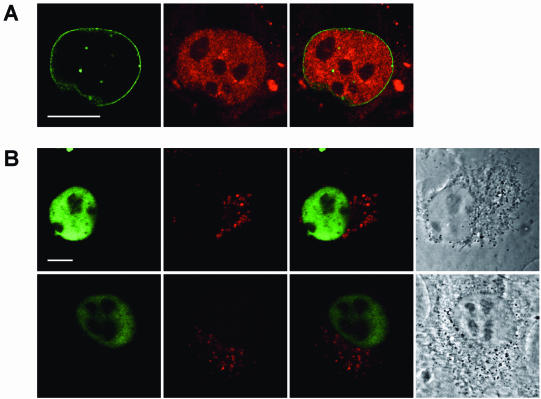
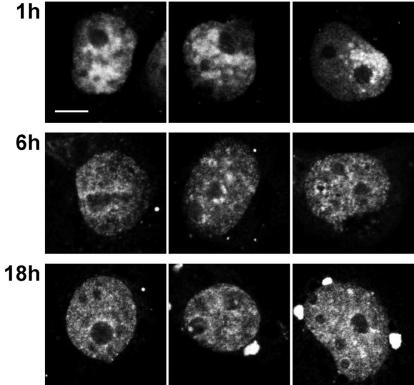
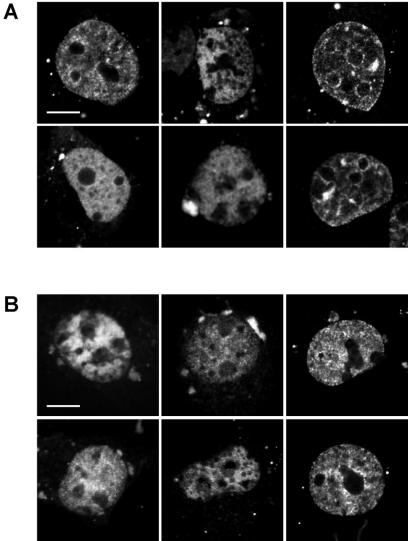
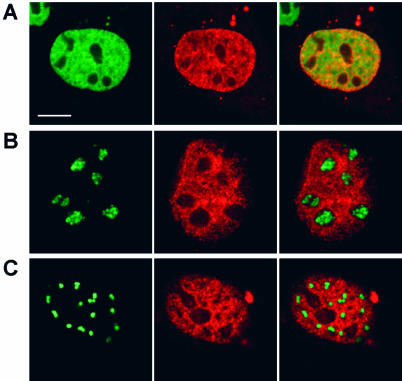
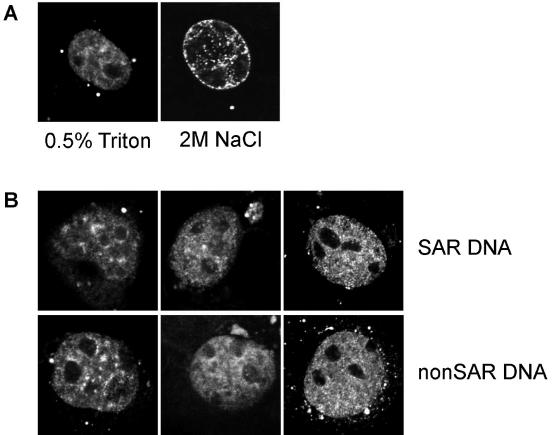
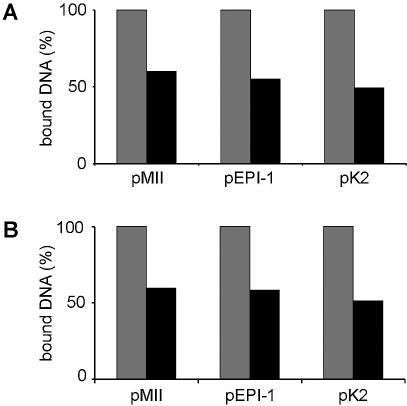
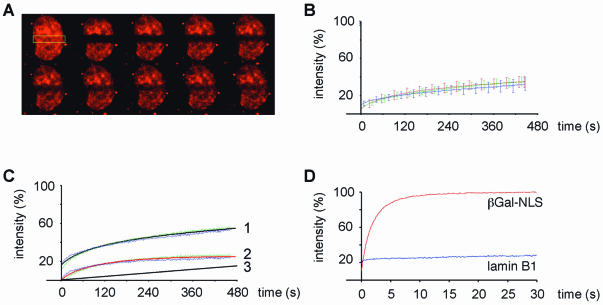
While genomic DNA, packaged into chromatin, is known to be locally constrained but highly dynamic in the nuclei of living cells, little is known about the localization and dynamics of small circular DNA molecules that invade cells by virus infection, application of gene therapy vectors or experimental transfection. To address this point, we have created traceable model substrates by direct labeling of plasmid DNA with fluorescent peptide nucleic acids, and have investigated their fate after microinjection into living cells. Here, we report that foreign DNA rapidly undergoes interactions with intranuclear structural sites that strongly reduce its mobility and restrict the DNA to regions excluding nucleoli and nuclear bodies such as PML bodies. The labeled plasmids partially co-localize with SAF-A, a well characterized marker protein for the nuclear 'scaffold' or 'matrix', and are resistant towards extraction by detergent and, in part, elevated salt concentrations. We show that the localization and the low mobility of plasmids is independent of the plasmid sequence, and does not require the presence of either a scaffold attachment region (SAR) DNA element or a functional promoter.
Figures
Similar articles
-
Can nuclear localization signals enhance nuclear localization of plasmid DNA?Bioconjug Chem. 2003 Mar-Apr;14(2):282-6. doi: 10.1021/bc025602h. Bioconjug Chem. 2003. PMID: 12643737
-
No enhancement of nuclear entry by direct conjugation of a nuclear localization signal peptide to linearized DNA.Bioconjug Chem. 2003 Nov-Dec;14(6):1197-202. doi: 10.1021/bc034075e. Bioconjug Chem. 2003. PMID: 14624635
-
Nuclear-associated plasmid, but not cell-associated plasmid, is correlated with transgene expression in cultured mammalian cells.Mol Ther. 2000 Apr;1(4):339-46. doi: 10.1006/mthe.2000.0054. Mol Ther. 2000. PMID: 10933952
-
Shear-induced degradation of plasmid DNA.J Pharm Sci. 2002 Jul;91(7):1581-9. doi: 10.1002/jps.10140. J Pharm Sci. 2002. PMID: 12115820 Review.
-
Nuclear dynamics: where genes are and how they got there.Genome Biol. 2001;2(3):REVIEWS0002. doi: 10.1186/gb-2001-2-3-reviews0002. Epub 2001 Mar 9. Genome Biol. 2001. PMID: 11276427 Free PMC article. Review.
Cited by
-
Characterization of exogenous DNA mobility in live cells through fluctuation correlation spectroscopy.Sci Rep. 2015 Sep 10;5:13848. doi: 10.1038/srep13848. Sci Rep. 2015. PMID: 26354725 Free PMC article.
-
Tracking of microinjected DNA in live cells reveals the intracellular behavior and elimination of extrachromosomal genetic material.Nucleic Acids Res. 2005 Nov 3;33(19):6296-307. doi: 10.1093/nar/gki946. Print 2005. Nucleic Acids Res. 2005. PMID: 16269822 Free PMC article.
-
Similar active genes cluster in specialized transcription factories.J Cell Biol. 2008 May 19;181(4):615-23. doi: 10.1083/jcb.200710053. J Cell Biol. 2008. PMID: 18490511 Free PMC article.
-
Intranuclear trafficking of episomal DNA is transcription-dependent.Mol Ther. 2007 Dec;15(12):2132-9. doi: 10.1038/sj.mt.6300275. Epub 2007 Jul 31. Mol Ther. 2007. PMID: 17667946 Free PMC article.
-
Tethered particle analysis of supercoiled circular DNA using peptide nucleic acid handles.Nat Protoc. 2014 Sep;9(9):2206-23. doi: 10.1038/nprot.2014.152. Epub 2014 Aug 21. Nat Protoc. 2014. PMID: 25144271
References
-
- Bode J., Goetze,S., Ernst,E., Huesemann,Y., Baer,A., Seibler,J. and Mielke,C. (2003) In Bernardi,G. (ed.), Gene Transfer and Expression in Mammalian Cells. Elsevier, Amsterdam, Vol. 38, pp. 551–572.
-
- Bode J., Benham,C., Knopp,A. and Mielke,C. (2000) Transcriptional augmentation: modulation of gene expression by scaffold/matrix-attached regions (S/MAR elements). Crit. Rev. Eukaryot. Gene Expr., 10, 73–90. - PubMed
-
- Mirkovitch J., Mirault,M.E. and Laemmli,U.K. (1984) Organization of the higher-order chromatin loop: specific DNA attachment sites on nuclear scaffold. Cell, 39, 223–232. - PubMed
-
- Cockerill P.N. and Garrard,W.T. (1986) Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell, 44, 273–282. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
