

Scoring profile-to-profile sequence alignments
- PMID: 15152092
- PMCID: PMC2279992
- DOI: 10.1110/ps.03601504
Scoring profile-to-profile sequence alignments
Abstract
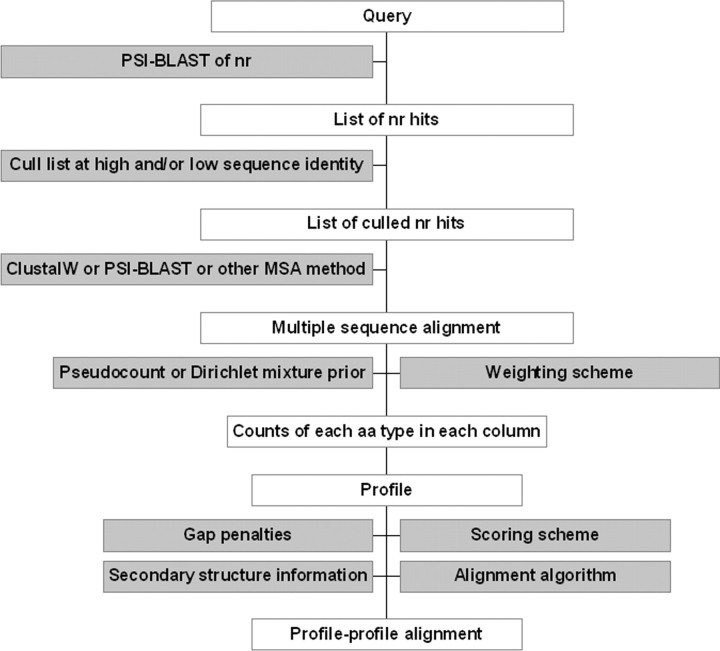
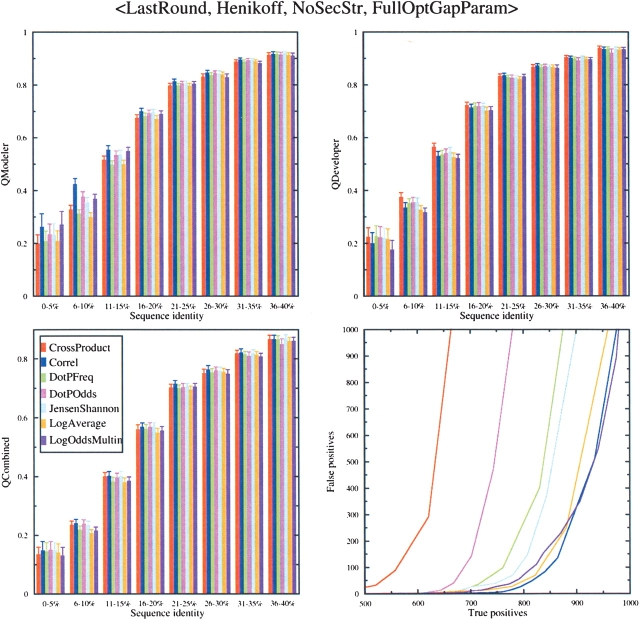
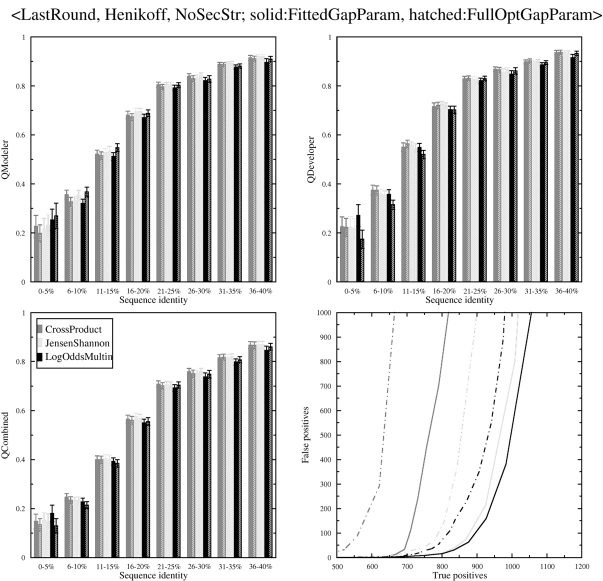
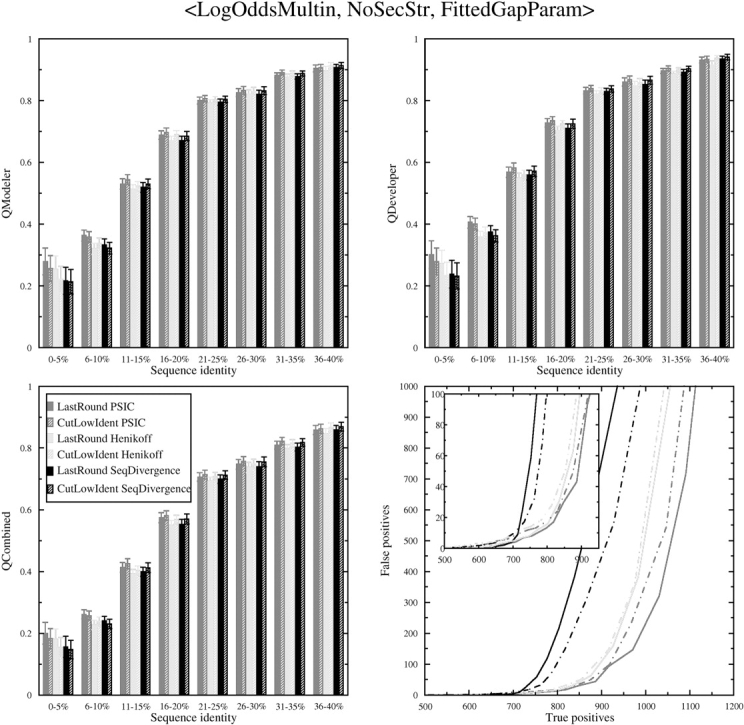
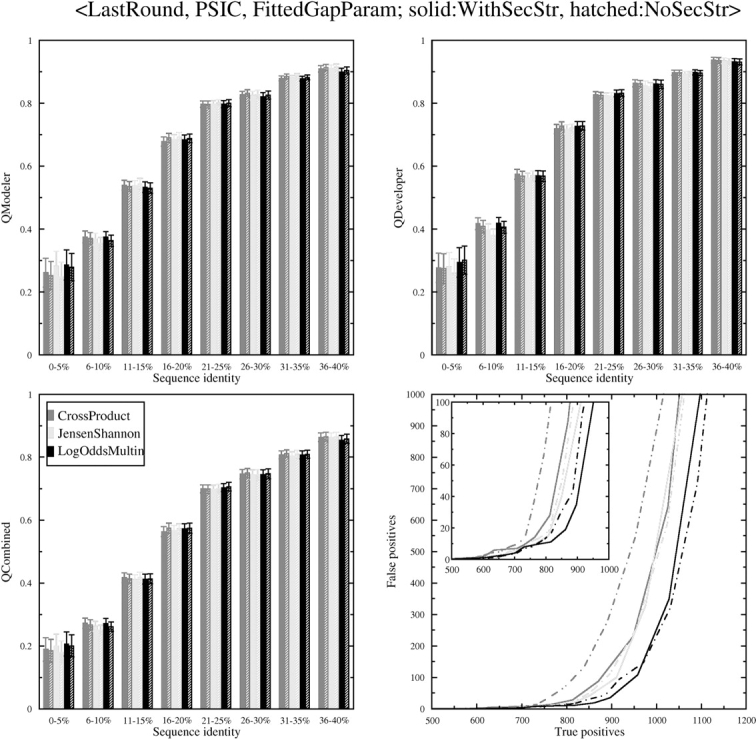
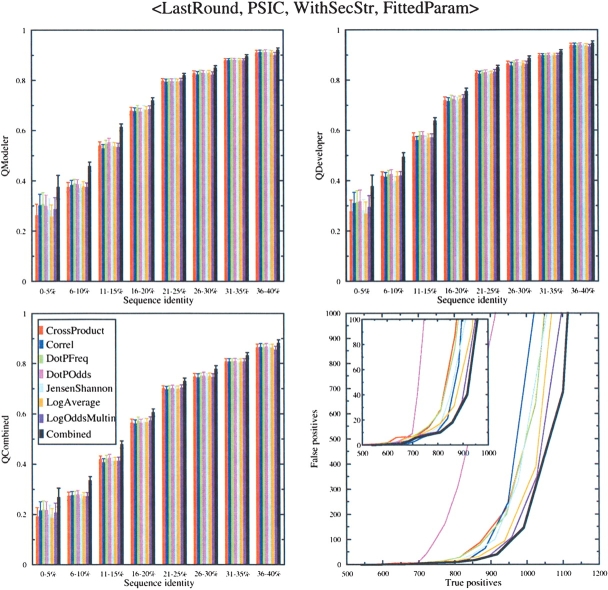
Sequence alignment profiles have been shown to be very powerful in creating accurate sequence alignments. Profiles are often used to search a sequence database with a local alignment algorithm. More accurate and longer alignments have been obtained with profile-to-profile comparison. There are several steps that must be performed in creating profile-profile alignments, and each involves choices in parameters and algorithms. These steps include (1) what sequences to include in a multiple alignment used to build each profile, (2) how to weight similar sequences in the multiple alignment and how to determine amino acid frequencies from the weighted alignment, (3) how to score a column from one profile aligned to a column of the other profile, (4) how to score gaps in the profile-profile alignment, and (5) how to include structural information. Large-scale benchmarks consisting of pairs of homologous proteins with structurally determined sequence alignments are necessary for evaluating the efficacy of each scoring scheme. With such a benchmark, we have investigated the properties of profile-profile alignments and found that (1) with optimized gap penalties, most column-column scoring functions behave similarly to one another in alignment accuracy; (2) some functions, however, have much higher search sensitivity and specificity; (3) position-specific weighting schemes in determining amino acid counts in columns of multiple sequence alignments are better than sequence-specific schemes; (4) removing positions in the profile with gaps in the query sequence results in better alignments; and (5) adding predicted and known secondary structure information improves alignments.
Figures
References
-
- Bourne, P.E. 2003. CASP and CAFASP experiments and their findings. Methods Biochem. Anal. 44 501–507. - PubMed
-
- Fischer, D., Elofsson, A., Rice, D., and Eisenberg, D. 1996. Assessing the performance of fold recognition methods by means of a comprehensive benchmark. Pac. Symp. Biocomput. pp. 300–318. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
