

Some fundamental aspects of building protein structures from fragment libraries
- PMID: 15152094
- PMCID: PMC2279988
- DOI: 10.1110/ps.03494504
Some fundamental aspects of building protein structures from fragment libraries
Abstract
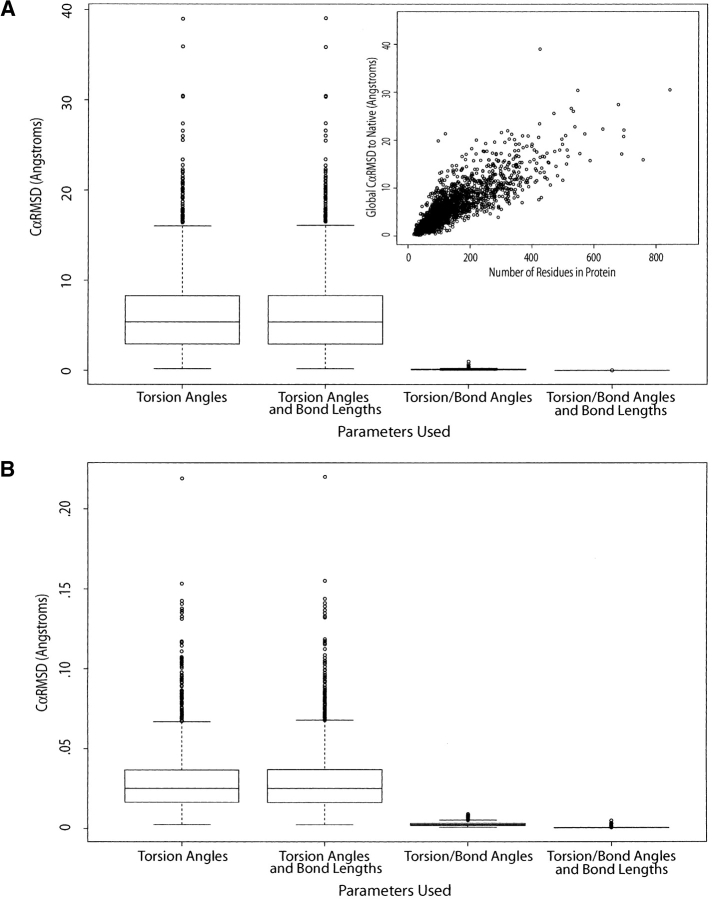
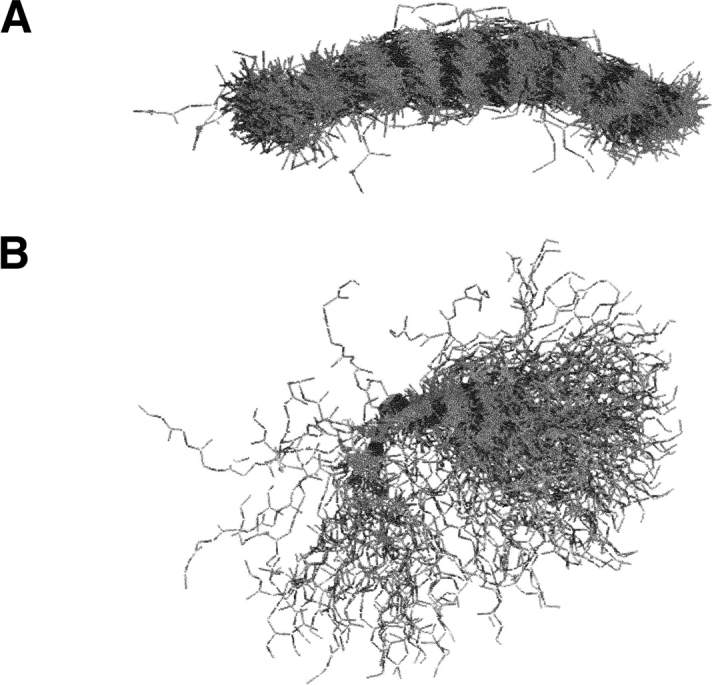
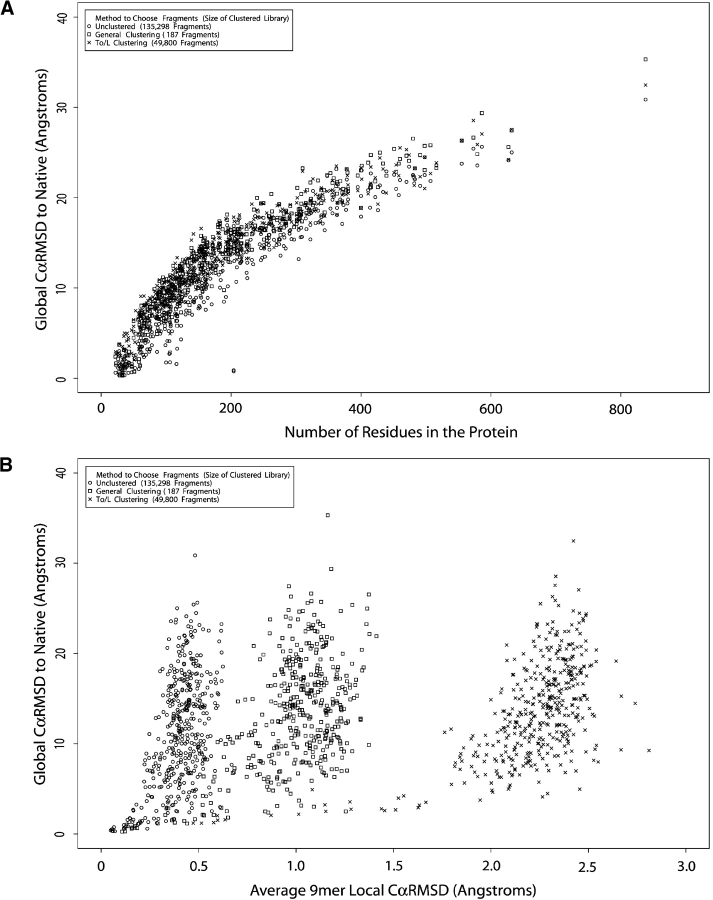
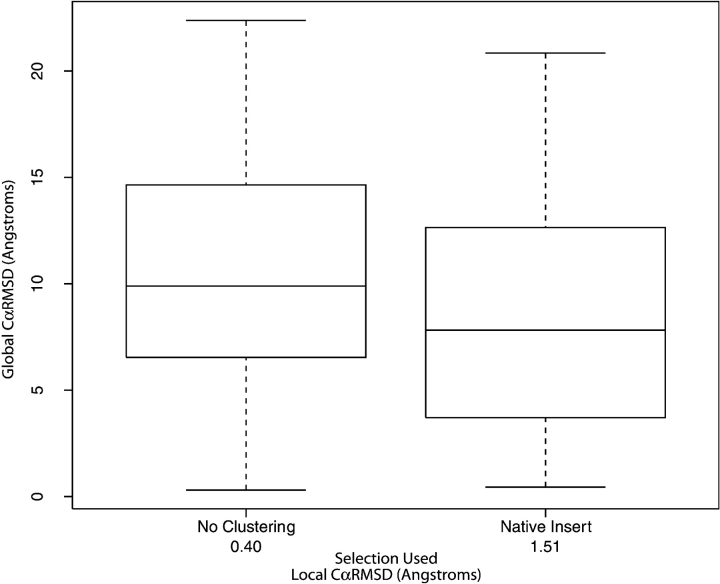
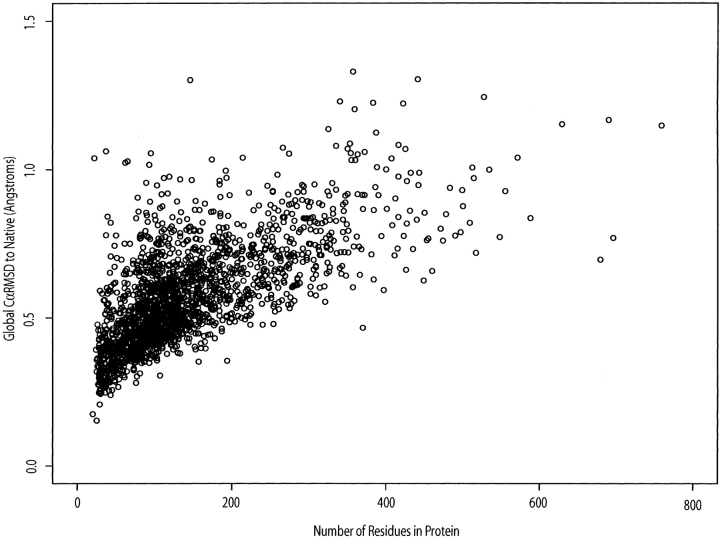
We have investigated some of the basic principles that influence generation of protein structures using a fragment-based, random insertion method. We tested buildup methods and fragment library quality for accuracy in constructing a set of known structures. The parameters most influential in the construction procedure are bond and torsion angles with minor inaccuracies in bond angles alone causing >6 A CalphaRMSD for a 150-residue protein. Idealization to a standard set of values corrects this problem, but changes the torsion angles and does not work for every structure. Alternatively, we found using Cartesian coordinates instead of torsion angles did not reduce performance and can potentially increase speed and accuracy. Under conditions simulating ab initio structure prediction, fragment library quality can be suboptimal and still produce near-native structures. Using various clustering criteria, we created a number of libraries and used them to predict a set of native structures based on nonnative fragments. Local CalphaRMSD fit of fragments, library size, and takeoff/landing angle criteria weakly influence the accuracy of the models. Based on a fragment's minimal perturbation upon insertion into a known structure, a seminative fragment library was created that produced more accurate structures with fragments that were less similar to native fragments than the other sets. These results suggest that fragments need only contain native-like subsections, which when correctly overlapped, can recreate a native-like model. For fragment-based, random insertion methods used in protein structure prediction and design, our findings help to define the parameters this method needs to generate near-native structures.
Figures
Similar articles
-
Protein structure prediction based on fragment assembly and parameter optimization.Biophys Chem. 2005 Apr 1;115(2-3):209-14. doi: 10.1016/j.bpc.2004.12.046. Epub 2005 Jan 6. Biophys Chem. 2005. PMID: 15752606
-
Small libraries of protein fragments model native protein structures accurately.J Mol Biol. 2002 Oct 18;323(2):297-307. doi: 10.1016/s0022-2836(02)00942-7. J Mol Biol. 2002. PMID: 12381322
-
Development of an ab initio protein structure prediction system ABLE.Genome Inform. 2003;14:228-37. Genome Inform. 2003. PMID: 15706537
-
[A turning point in the knowledge of the structure-function-activity relations of elastin].J Soc Biol. 2001;195(2):181-93. J Soc Biol. 2001. PMID: 11727705 Review. French.
-
Protein design with fragment databases.Curr Opin Struct Biol. 2011 Aug;21(4):452-9. doi: 10.1016/j.sbi.2011.05.002. Epub 2011 Jun 16. Curr Opin Struct Biol. 2011. PMID: 21684149 Review.
Cited by
-
Building a better fragment library for de novo protein structure prediction.PLoS One. 2015 Apr 22;10(4):e0123998. doi: 10.1371/journal.pone.0123998. eCollection 2015. PLoS One. 2015. PMID: 25901595 Free PMC article.
-
Near-native protein loop sampling using nonparametric density estimation accommodating sparcity.PLoS Comput Biol. 2011 Oct;7(10):e1002234. doi: 10.1371/journal.pcbi.1002234. Epub 2011 Oct 20. PLoS Comput Biol. 2011. PMID: 22028638 Free PMC article.
-
Characterization and Prediction of Protein Flexibility Based on Structural Alphabets.Biomed Res Int. 2016;2016:4628025. doi: 10.1155/2016/4628025. Epub 2016 Aug 30. Biomed Res Int. 2016. PMID: 27660756 Free PMC article.
-
Critical Features of Fragment Libraries for Protein Structure Prediction.PLoS One. 2017 Jan 13;12(1):e0170131. doi: 10.1371/journal.pone.0170131. eCollection 2017. PLoS One. 2017. PMID: 28085928 Free PMC article.
-
Building native protein conformation from highly approximate backbone torsion angles.Proc Natl Acad Sci U S A. 2005 Nov 8;102(45):16227-32. doi: 10.1073/pnas.0508415102. Epub 2005 Oct 26. Proc Natl Acad Sci U S A. 2005. PMID: 16251268 Free PMC article.
References
-
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S., et al. 2002. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58 899–907. - PubMed
-
- Bonneau, R. and Baker, D. 2001. Ab initio protein structure prediction: Progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 30 173–189. - PubMed
-
- Bonneau, R., Tsai, J., Ruczinski, I., Chivian, D., Rohl, C., Strauss, C.E., and Baker, D. 2001. Rosetta in CASP4: Progress in ab initio protein structure prediction. Proteins Suppl. 5: 119–126. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
