

High prevalence of SLC6A8 deficiency in X-linked mental retardation
- PMID: 15154114
- PMCID: PMC1182013
- DOI: 10.1086/422102
High prevalence of SLC6A8 deficiency in X-linked mental retardation
Abstract
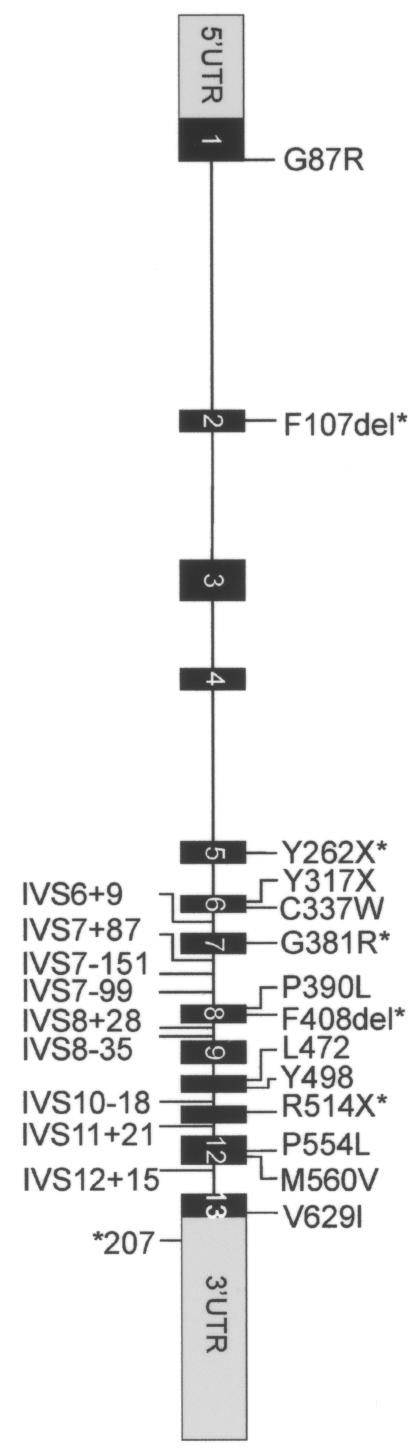
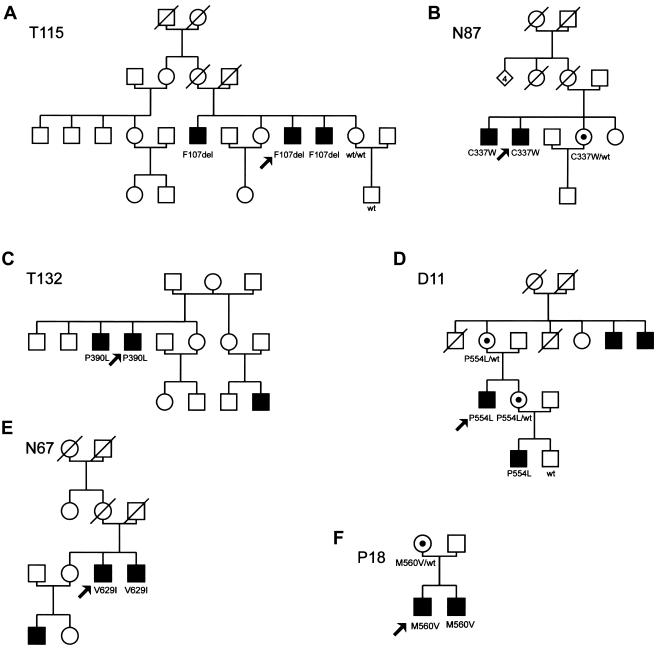
A novel X-linked mental retardation (XLMR) syndrome was recently identified, resulting from creatine deficiency in the brain caused by mutations in the creatine transporter gene, SLC6A8. We have studied the prevalence of SLC6A8 mutations in a panel of 290 patients with nonsyndromic XLMR archived by the European XLMR Consortium. The full-length open reading frame and splice sites of the SLC6A8 gene were investigated by DNA sequence analysis. Six pathogenic mutations, of which five were novel, were identified in a total of 288 patients with XLMR, showing a prevalence of at least 2.1% (6/288). The novel pathogenic mutations are a nonsense mutation (p.Y317X) and four missense mutations. Three missense mutations (p.G87R, p.P390L, and p.P554L) were concluded to be pathogenic on the basis of conservation, segregation, chemical properties of the residues involved, as well as the absence of these and any other missense mutation in 276 controls. For the p.C337W mutation, additional material was available to biochemically prove (i.e., by increased urinary creatine : creatinine ratio) pathogenicity. In addition, we found nine novel polymorphisms (IVS1+26G-->A, IVS7+37G-->A, IVS7+87A-->G, IVS7-35G-->A, IVS12-3C-->T, IVS2+88G-->C, IVS9-36G-->A, IVS12-82G-->C, and p.Y498) that were present in the XLMR panel and/or in the control panel. Two missense variants (p.V629I and p.M560V) that were not highly conserved and were not associated with increased creatine : creatinine ratio, one translational silent variant (p.L472), and 10 intervening sequence variants or untranslated region variants (IVS6+9C-->T, IVS7-151_152delGA, IVS7-99C-->A, IVS8-35G-->A, IVS8+28C-->T, IVS10-18C-->T, IVS11+21G-->A, IVS12+15C-->T, *207G-->C, IVS12+32C-->A) were found only in the XLMR panel but should be considered as unclassified variants or as a polymorphism (p.M560V). Our data indicate that the frequency of SLC6A8 mutations in the XLMR population is close to that of CGG expansions in FMR1, the gene responsible for fragile-X syndrome.
Figures


Comment in
-
Comparative frequency of fragile-X (FMR1) and creatine transporter (SLC6A8) mutations in X-linked mental retardation.Am J Hum Genet. 2004 Oct;75(4):730-1; author reply 731-2. doi: 10.1086/424821. Am J Hum Genet. 2004. PMID: 15338463 Free PMC article. No abstract available.
References
Electronic-Database Information
-
- European XLMR Consortium, http://www.euromrx.com/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for SLC6A8 [accession number Z66539])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for SLC6A8, AGAT deficiency, GAMT deficiency, and SLC6A8 deficiency)
References
-
- Brown AN, Muth TR, Caplan MJ (2003) The C-terminal tail of the GAT-2 GABA transporter contains a novel motif that plays a role in basolateral targeting. Am J Physiol Cell Physiol 286:c1071–c1077 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
