

Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets
- PMID: 15157079
- PMCID: PMC2891762
- DOI: 10.1021/bi0496945
Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets
Abstract
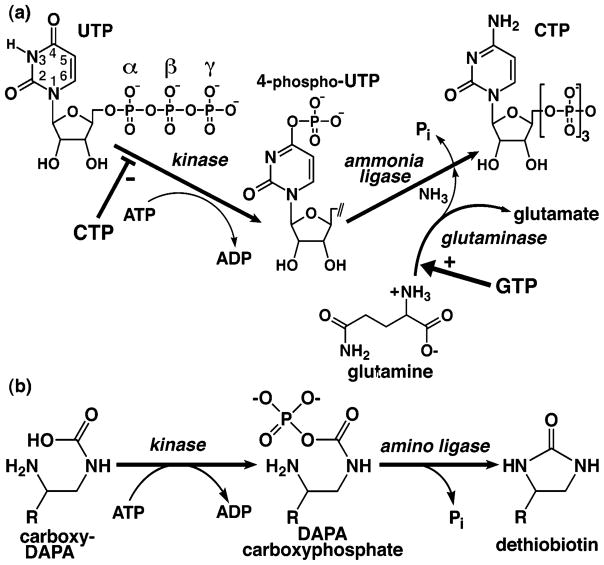
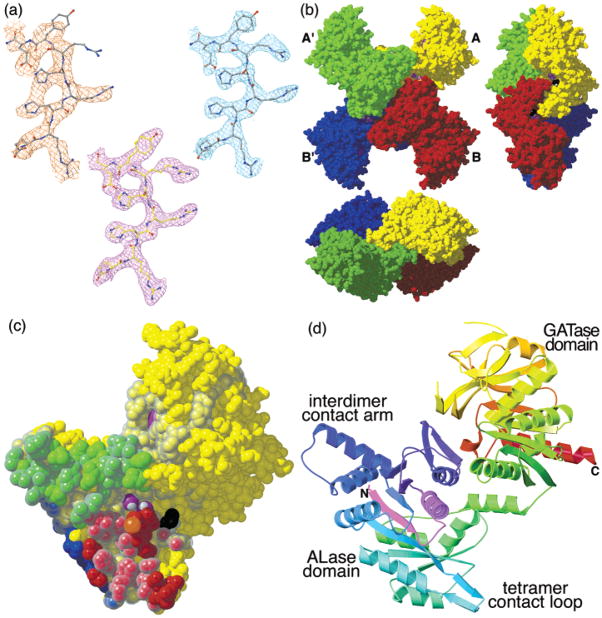
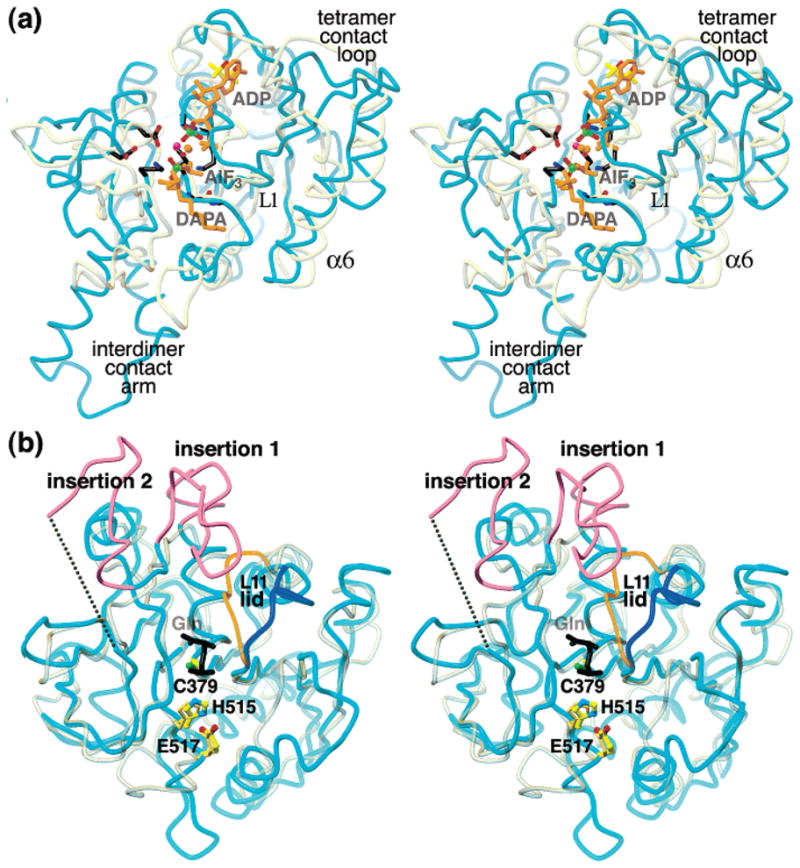
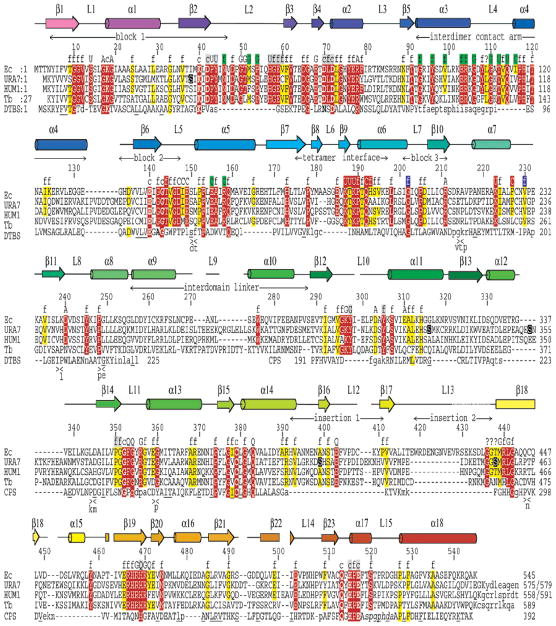
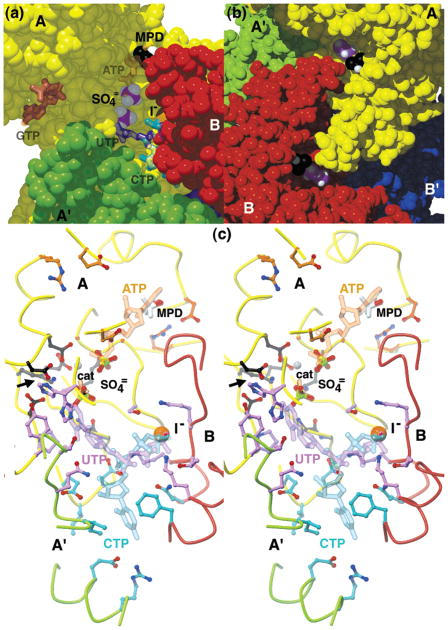
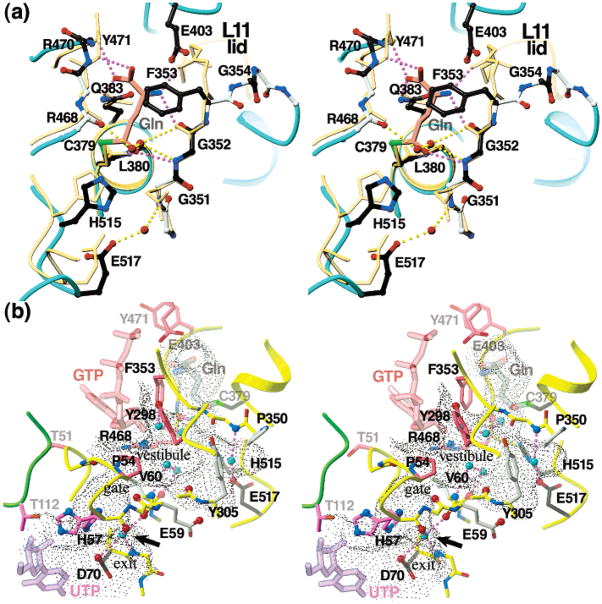
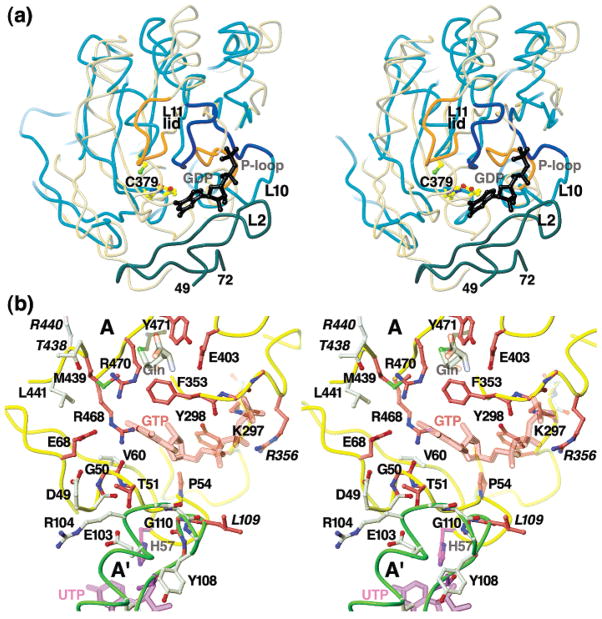
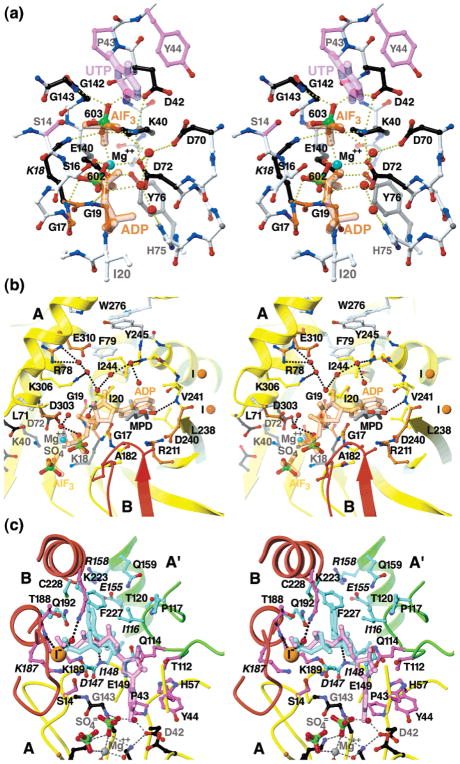
Cytidine triphosphate synthetases (CTPSs) produce CTP from UTP and glutamine, and regulate intracellular CTP levels through interactions with the four ribonucleotide triphosphates. We solved the 2.3-A resolution crystal structure of Escherichia coli CTPS using Hg-MAD phasing. The structure reveals a nearly symmetric 222 tetramer, in which each bifunctional monomer contains a dethiobiotin synthetase-like amidoligase N-terminal domain and a Type 1 glutamine amidotransferase C-terminal domain. For each amidoligase active site, essential ATP- and UTP-binding surfaces are contributed by three monomers, suggesting that activity requires tetramer formation, and that a nucleotide-dependent dimer-tetramer equilibrium contributes to the observed positive cooperativity. A gated channel that spans 25 A between the glutamine hydrolysis and amidoligase active sites provides a path for ammonia diffusion. The channel is accessible to solvent at the base of a cleft adjoining the glutamine hydrolysis active site, providing an entry point for exogenous ammonia. Guanine nucleotide binding sites of structurally related GTPases superimpose on this cleft, providing insights into allosteric regulation by GTP. Mutations that confer nucleoside drug resistance and release CTP inhibition map to a pocket that neighbors the UTP-binding site and can accommodate a pyrimidine ring. Its location suggests that competitive feedback inhibition is affected via a distinct product/drug binding site that overlaps the substrate triphosphate binding site. Overall, the E. coli structure provides a framework for homology modeling of other CTPSs and structure-based design of anti-CTPS therapeutics.
Figures
Similar articles
-
Limited proteolysis of Escherichia coli cytidine 5'-triphosphate synthase. Identification of residues required for CTP formation and GTP-dependent activation of glutamine hydrolysis.Eur J Biochem. 2003 May;270(10):2195-206. doi: 10.1046/j.1432-1033.2003.03588.x. Eur J Biochem. 2003. PMID: 12752439
-
Mechanisms of product feedback regulation and drug resistance in cytidine triphosphate synthetases from the structure of a CTP-inhibited complex.Biochemistry. 2005 Oct 18;44(41):13491-9. doi: 10.1021/bi051282o. Biochemistry. 2005. PMID: 16216072 Free PMC article.
-
Aspartate-107 and leucine-109 facilitate efficient coupling of glutamine hydrolysis to CTP synthesis by Escherichia coli CTP synthase.Biochem J. 2003 Feb 1;369(Pt 3):497-507. doi: 10.1042/BJ20021110. Biochem J. 2003. PMID: 12383057 Free PMC article.
-
GTP-Dependent Regulation of CTP Synthase: Evolving Insights into Allosteric Activation and NH3 Translocation.Biomolecules. 2022 Apr 29;12(5):647. doi: 10.3390/biom12050647. Biomolecules. 2022. PMID: 35625575 Free PMC article. Review.
-
The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia.Biochemistry. 1999 Jun 22;38(25):7891-9. doi: 10.1021/bi990871p. Biochemistry. 1999. PMID: 10387030 Review.
Cited by
-
Human CTP synthase filament structure reveals the active enzyme conformation.Nat Struct Mol Biol. 2017 Jun;24(6):507-514. doi: 10.1038/nsmb.3407. Epub 2017 May 1. Nat Struct Mol Biol. 2017. PMID: 28459447 Free PMC article.
-
Proteomic Analysis Reveals a Biofilm-Like Behavior of Planktonic Aggregates of Staphylococcus epidermidis Grown Under Environmental Pressure/Stress.Front Microbiol. 2019 Sep 6;10:1909. doi: 10.3389/fmicb.2019.01909. eCollection 2019. Front Microbiol. 2019. PMID: 31551940 Free PMC article.
-
Structure of the synthetase domain of human CTP synthetase, a target for anticancer therapy.Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006 Jul 1;62(Pt 7):613-7. doi: 10.1107/S1744309106018136. Epub 2006 Jun 10. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006. PMID: 16820675 Free PMC article.
-
Regulation of human cytidine triphosphate synthetase 2 by phosphorylation.J Biol Chem. 2010 Oct 29;285(44):33727-36. doi: 10.1074/jbc.M110.178566. Epub 2010 Aug 25. J Biol Chem. 2010. PMID: 20739275 Free PMC article.
-
CTP synthase: the hissing of the cellular serpent.Histochem Cell Biol. 2022 Dec;158(6):517-534. doi: 10.1007/s00418-022-02133-w. Epub 2022 Jul 26. Histochem Cell Biol. 2022. PMID: 35881195 Free PMC article. Review.
References
-
- Liebermann I. Enzymatic Amination of uridine triphosphate to cytidine triphosphate. J Am Chem Soc. 1955;55:2661–2662. - PubMed
-
- Jones ME. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem. 1980;49:253–279. - PubMed
-
- Kent C, Carman GM, Spence MW, Dowhan W. Regulation of eukaryotic phospholipid metabolism. FASEB J. 1991;5:2258–2266. - PubMed
-
- Kent C, Carman GM. Interactions among pathways for phosphatidylcholine metabolism, CTP synthesis and secretion through the Golgi apparatus. Trends Biochem Sci. 1999;24:146–150. - PubMed
-
- West TP, O’Donovan GA. Repression of cytidine triphosphate synthetase in Salmonella typhimurium by pyrimidines during uridine nucleotide depletion. J Gen Microbiol. 1982;128:895–899. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
