

Infectious agents and age-related neurodegenerative disorders
- PMID: 15163105
- PMCID: PMC7172323
- DOI: 10.1016/j.arr.2003.08.005
Infectious agents and age-related neurodegenerative disorders
Erratum in
- Ageing Res Rev. 2004 Apr;3(2):349
Abstract
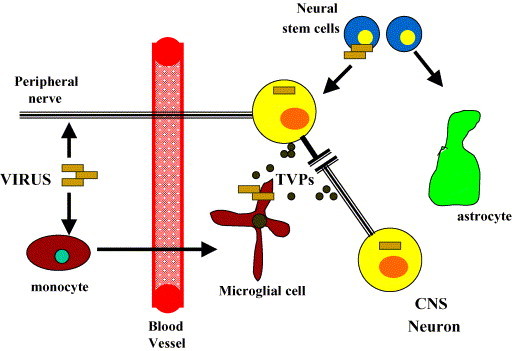
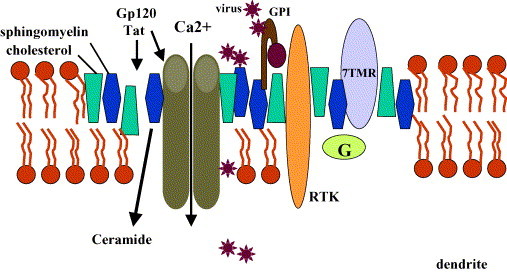
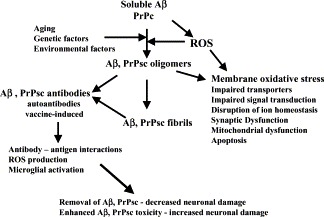
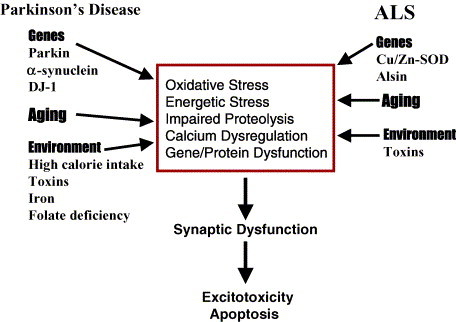
chlamdAs with other organ systems, the vulnerability of the nervous system to infectious agents increases with aging. Several different infectious agents can cause neurodegenerative conditions, with prominent examples being human immunodeficiency virus (HIV-1) dementia and prion disorders. Such infections of the central nervous system (CNS) typically have a relatively long incubation period and a chronic progressive course, and are therefore increasing in frequency as more people live longer. Infectious agents may enter the central nervous system in infected migratory macrophages, by transcytosis across blood-brain barrier cells or by intraneuronal transfer from peripheral nerves. Synapses and lipid rafts are important sites at which infectious agents may enter neurons and/or exert their cytotoxic effects. Recent findings suggest the possibility that infectious agents may increase the risk of common age-related neurodegenerative disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS) and stroke. While scenarios can be envisioned whereby viruses such as Chlamydia pneumoniae, herpes simplex and influenza promote damage to neurons during aging, there is no conclusive evidence for a major role of these pathogens in neurodegenerative disorders. In the case of stroke, blood vessels may be adversely affected by bacteria or viruses resulting in atherosclerosis.
Figures
References
-
- Antezana D.F., Clatterbuck R.E., Alkayed N.J., Murphy S.J., Anderson L.G., Frazier J., Hurn P.D., Traystman R.J., Tamargo R.J. High-dose ibuprofen for reduction of striatal infarcts during middle cerebral artery occlusion in rats. J. Neurosurg. 2003;98:860–866. - PubMed
-
- Balin B.J., Appelt D.M. Role of infection in Alzheimer’s disease. J. Am. Osteopath. Assoc. 2001;101:S1–6. - PubMed
-
- Balin B.J., Gerard H.C., Arking E.J., Appelt D.M., Branigan P.J., Abrams J.T., Whittum-Hudson J.A., Hudson A.P. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. (Berl.) 1998;187:23–42. - PubMed
-
- Berger J.R., Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. J. Psychopharmacol. 2000;14:214–221. - PubMed
-
- Bruce A.J., Boling W., Kindy M.S., Peschon J., Kraemer P.J., Carpenter M.K., Holtsberg F.W., Mattson M.P. Altered neuronal and microglial responses to brain injury in mice lacking TNF receptors. Nat. Med. 1996;2:788–794. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
