

Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor
- PMID: 15169883
- PMCID: PMC419881
- DOI: 10.1128/MCB.24.12.5172-5183.2004
Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor
Abstract
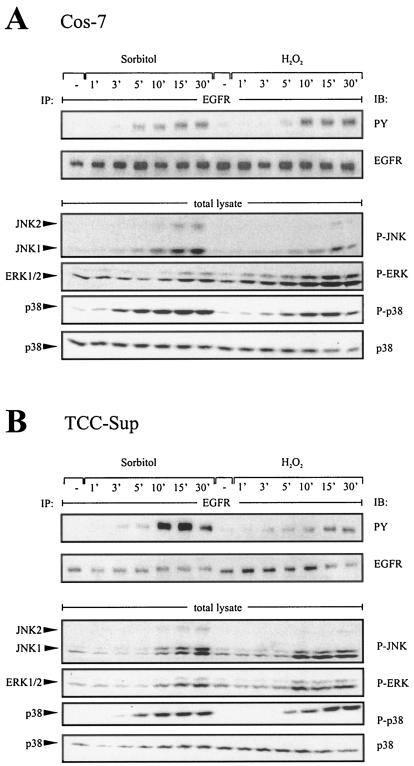
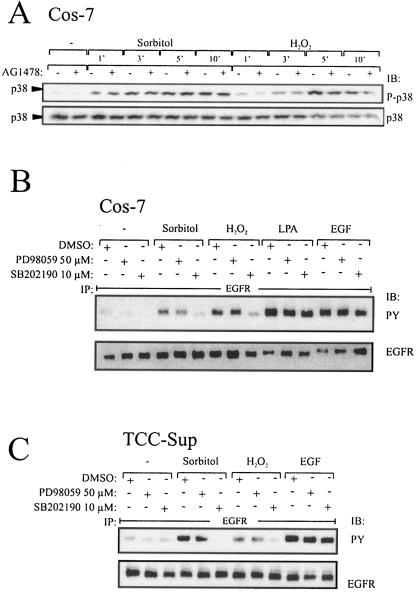
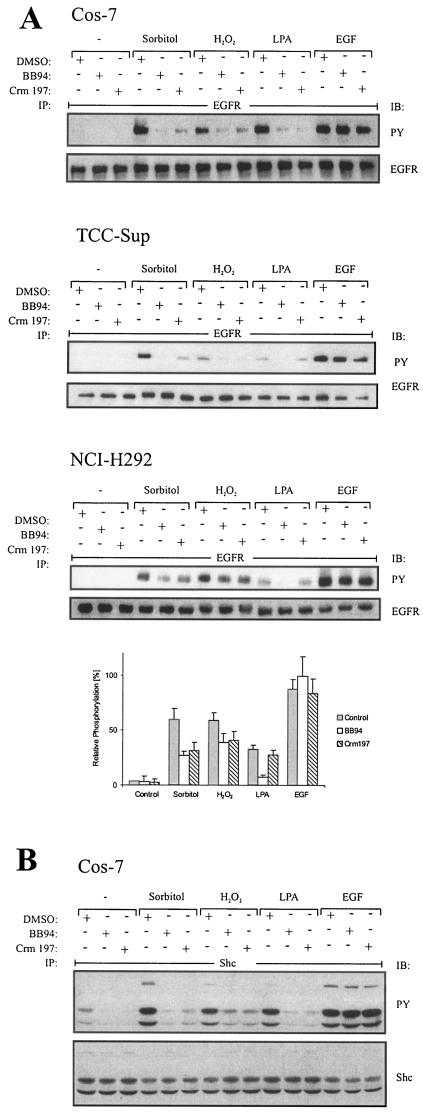
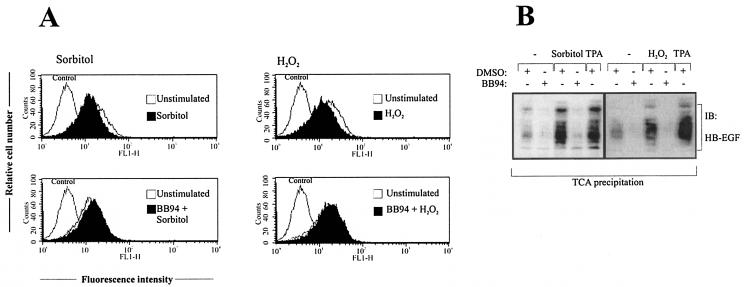
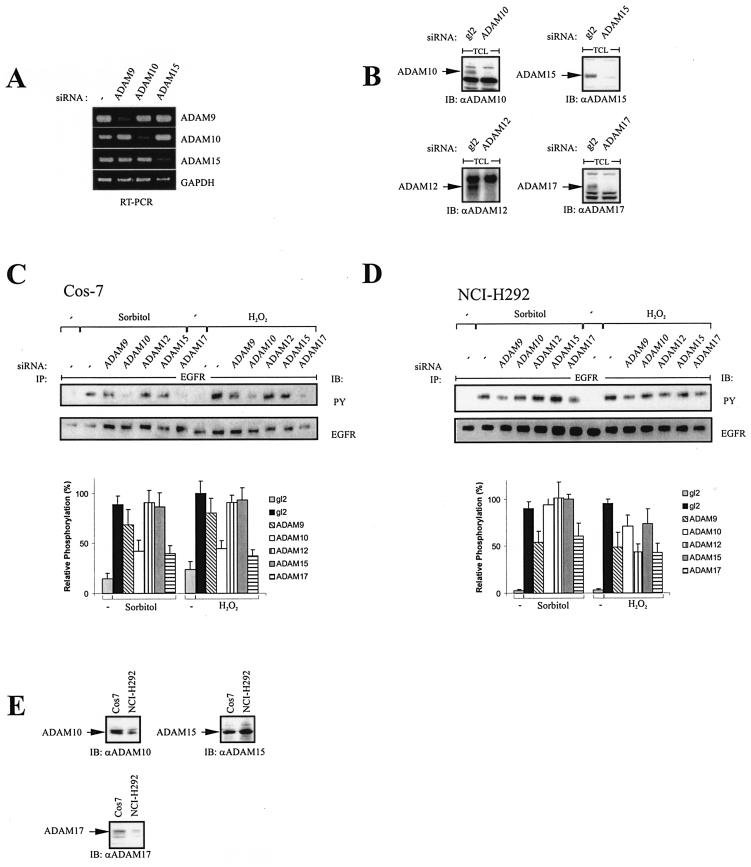
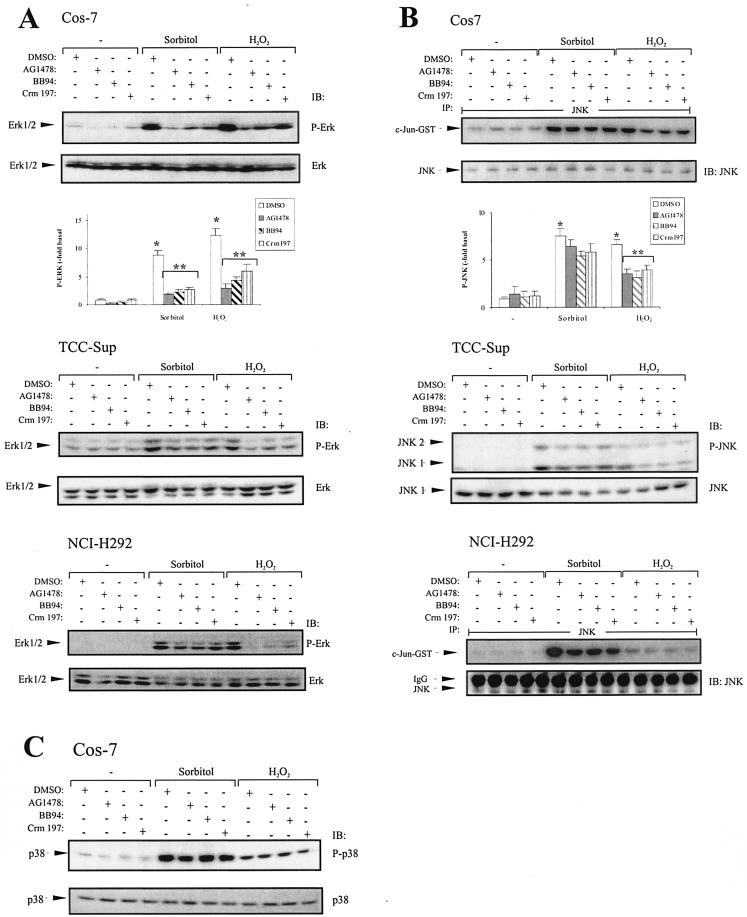
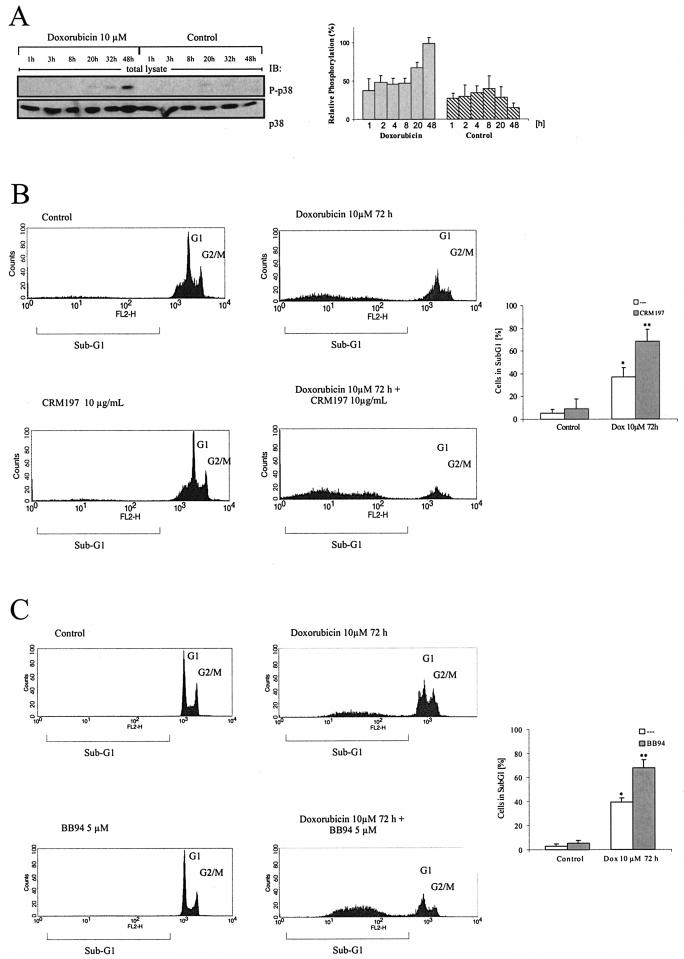
Mammalian cells respond to environmental stress by activating a variety of protein kinases critical for cellular signal transmission, such as the epidermal growth factor receptor (EGFR) tyrosine kinase and different members of the mitogen-activated protein kinase (MAPK) family. EGFR activation by stress stimuli was previously thought to occur independently of stimulation by extracellular ligands. Here, we provide evidence that osmotic and oxidative stresses induce a metalloprotease activity leading to cell surface cleavage of pro-heparin-binding EGF (pro-HB-EGF) and subsequent EGFR activation. This ligand-dependent EGFR signal resulted from stress-induced activation of the MAPK p38 in human carcinoma cells and was mediated by the metalloproteases ADAM9, -10, and -17. Furthermore, stress-induced EGFR activation induced downstream signaling through the MAPKs extracellular signal-regulated kinases 1 and 2 and JNK. Interestingly, apoptosis induced by treatment of tumor cells with doxorubicin was strongly enhanced by blocking HB-EGF function. Together, our data provide novel insights into the mammalian stress response, suggesting a broad mechanistic relevance of a p38-ADAM-HB-EGF-EGFR-dependent pathway and its potential significance for tumor cells in evasion of chemotherapeutic agent-induced apoptosis.
Figures
References
-
- Asakura, M., M. Kitakaze, S. Takashima, Y. Liao, F. Ishikura, T. Yoshinaka, H. Ohmoto, K. Node, K. Yoshino, H. Ishiguro, H. Asanuma, S. Sanada, Y. Matsumura, H. Takeda, S. Beppu, M. Tada, M. Hori, and S. Higashiyama. 2002. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat. Med. 8:35-40. - PubMed
-
- Biscardi, J. S., M. C. Maa, D. A. Tice, M. E. Cox, T. H. Leu, and S. J. Parsons. 1999. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 274:8335-8343. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous